一种Au‑ZrO2催化剂及其应用的利记博彩app
本发明涉及催化剂制备领域,具体涉及一种au-zro2催化剂及其应用。
背景技术:
水煤气反应(wgs)在获得洁净氢气的同时也消除了一氧化碳。近年来,以纯氢为燃料的质子交换膜燃料电池技术的兴起,使原本传统的变换催化剂不能满足燃料电池体系的要求。因此,开发具有高活性和稳定性的新型变换催化剂具有重要的意义。
上世纪八十年代,日本的haeuta发现负载在过渡金属氧化物上的金催化剂对co低温氧化具有很高的催化活性,负载au催化剂具有较高的低温活性、较宽的活性温区、较好的抗氧化性能以及抗水性能,因而被认为是最适合用于pemfc苛刻操作环境的wgs催化剂之一。
zro2具有酸碱性、氧化还原性、良好的热稳定性和机械强度,作为催化剂载体的研究日益受到人们的重视,得到较为广泛的应用。
例如,申请公布号为cn102389795a的发明专利申请公开了一种用于甲酸分解制氢的纳米金催化剂及其制备方法,该催化剂由载体和活性组份组成,载体为zro2或tio2,活性组份为纳米尺寸的金属态金,金的质量含量为0.45%~5%,分别记为au/zro2和au/tio2。该催化剂对于多相甲酸分解制氢,在环境温度下具有较高的活性和稳定性,并且具有制备方法简易、快捷、成本低廉等优点。
其中,au-zro2催化剂也已经被报道具有很高的wgs活性。
申请公布号为cn101954279a的发明专利申请公开了一种应用于富氢重整气下的低温水煤气变换催化剂及其制备方法,该催化剂的组成为au/zro2,其中活性组分为au,载体为zro2,au的重量含量为0.1~3%,zro2的重量含量为97~99.9%。该方法制备的zro2载体具有较高的比表面积和较大的孔体积,可更好的分散活性组分au。本发明中的催化剂在金质量含量较低的情况下依然具有很高的低温水煤气变换催化活性,如:au重量含量降为1%的au/zro2催化剂,在210~300℃反应温区内的co转化率仍接近平衡转化率。
技术实现要素:
本发明提供了一种改性au-zro2催化剂以及其在水煤气变换反应制备氢气中的应用,该改性au-zro2催化剂具有较高的催化活性。
一种改性au-zro2催化剂,包括载体和活性组分,所述载体由zro2在氢气氛围下焙烧改性后得到。
其中,所述zro2采用水热法进行制备,具体为:将zrocl2·8h2o在150℃条件下水热反应6小时后,经洗涤、干燥后,得到zro2。
经研究发现,在氢气气氛下处理zro2能有效提升催化剂的催化活性,综合表征结果发现,改性au-zro2催化剂的活性与其比表面积、孔容积及zro2载体的晶粒大小和表面氧空位等息息相关。其中,氢气处理所致的表面氧空位有利于负载金以零价态au(非氧化态)的形式存在,从而表现出更好的催化活性。
所以,对于上述改性au-zro2催化剂,所述活性组分为单质金。
进一步地,所述载体为双介孔型zro2,包含3~6nm的介孔和30~40nm的堆砌孔,有利于提高载体和催化剂的比表面积,提高以氢气焙烧zro2为载体的催化剂活性。
进一步地,所述载体的焙烧条件为:在500~600℃下常压焙烧2~6h,或者,在200~300℃、10~20atm下焙烧2~4h。
进一步地,所述改性au-zro2催化剂的比表面积为20~30m2/g;平均孔隙半径为20~40nm。
进一步地,所述改性zro2载体和改性au-zro2催化剂的电子密度分别为1×1023~9×1023cm-3和1×1024~9×1024cm-3。
本发明还提供了所述改性au-zro2催化剂在水煤气变换反应制备氢气中的应用。
本发明提供了一种利用改性au-zro2催化剂进行水煤气变换反应制备氢气的方法,所述反应的温度为200~600℃。
水煤气变换反应体系中,水蒸汽与原料气的比例保持在0.5:1~1:1,原料气中co体积浓度为1~15%,h2体积浓度为0~50%,co2体积浓度为0~12%。在上述反应条件更为严苛的情况下,本发明催化剂仍能显著提高co转化率。
与现有技术相比,本发明具有以下有益效果:
本发明将水热法制备的zro2置于氢气氛围下进行焙烧获得改性载体,再负载金,使获得的改性催化剂的比表面积提高、平均孔径减少、表面氧空位明显增大,并使负载金以零价态au(非氧化态)的形式存在,抑制了电子-空穴复合,增加了供体密度的同时提升了电荷传输效率,从而显著提高催化剂的催化活性,提高了水煤气变换反应中co的转化率。
附图说明
图1为实施例1和对比例1-7制备的改性au-zro2催化剂的wgs反应催化活性结果。
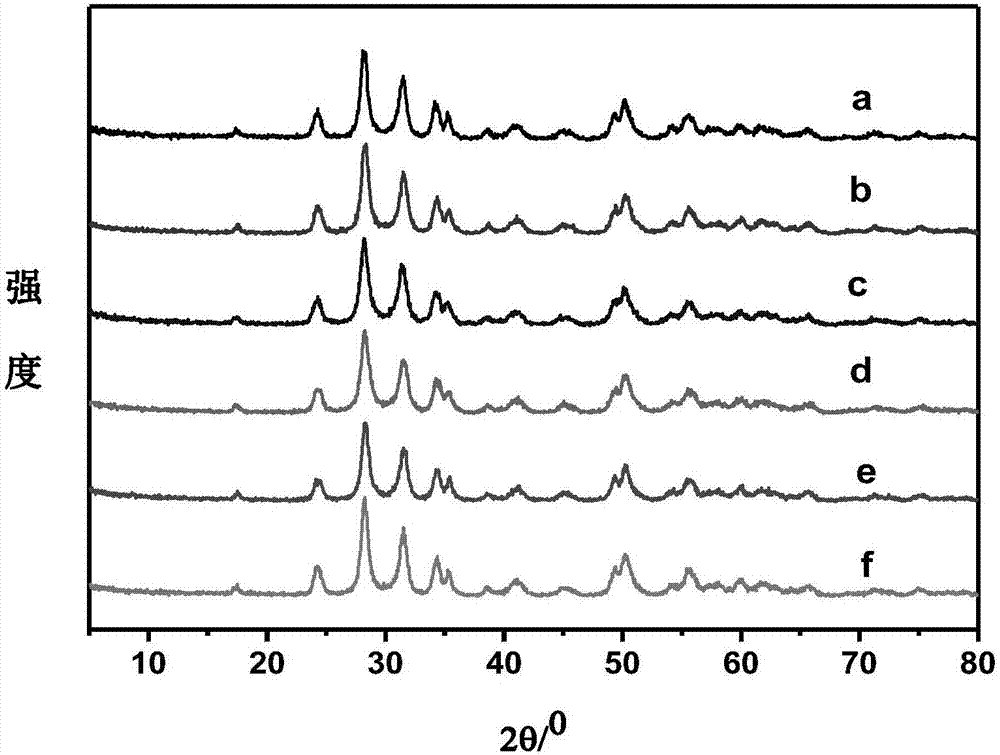
图2为实施例1和对比例1、2制备的改性zro2载体和改性au-zro2催化剂的xrd图谱;
其中,a:au-zro2-ht-a;b:zro2-ht-a;c:au-zro2-ht-h;d:zro2-ht-h;e:au-zro2-ht-a-h;f:zro2-ht-a-h。
图3为实施例1和对比例1制备的改性au-zro2催化剂的sem照片结果;
其中,a:au-zro2-ht-a;b:au-zro2-ht-h。
图4为实施例1和对比例1、2制备的改性zro2载体和改性au-zro2催化剂的tem图和hrtem(虚线是无序层和结晶核的界面)图;
其中,a、b和g依次为au-zro2-ht-h、au-zro2-ht-a-h和au-zro2-ht-a的tem图;c、e、h、d、f和i依次为zro2-ht-h、zro2-ht-a-h、zro2-ht-a、au-zro2-ht-h、au-zro2-ht-a-h和au-zro2-ht-a的hrtem图。
图5为实施例1和对比例1、2制备的改性zro2载体和改性au-zro2催化剂的n2物理吸脱附曲线。
图6为实施例1和对比例1、2制备的改性zro2载体和改性au-zro2催化剂的孔径分布图。
图7为实施例1和对比例1、2制备的改性zro2载体和改性au-zro2催化剂的xps光谱图。
图8为实施例1和对比例1、2制备的改性zro2载体和改性au-zro2催化剂的epr谱图。
图9为实施例1和对比例1、2制备的改性zro2载体和改性au-zro2催化剂的荧光光谱图。
图10为实施例1和对比例1、2制备的改性zro2载体和改性au-zro2催化剂的mott-schottky图。
具体实施方式
以下结合具体实施例对本发明做进一步地详细说明,但本发明并不仅限于此。本发明中未详细说明的测定方法均为本领域常规技术。
本发明实施例中采用的主要仪器:x-射线粉末衍射仪(x'pertpro)购自荷兰panalytic公司;拉曼光谱仪(renishawinviaplus)购自英国雷尼绍公司;比表面测试仪(micrometricsasap2020)购自美国麦克公司;x-射线光电子能谱仪(vgescalab250)购自美国热电公司;高分辨率透射电子显微镜(jeol2010f(200kv))购自日本jeol公司;场发射扫描电子显微镜(hitachis-4800)购自日本日立公司;电子顺磁共振仪(brukeremx-8)购于德国布鲁克公司;荧光光谱仪(hitachif-4500)购于日本日立公司;电化学工作站(chi660d)购于上海辰华仪器有限公司;气固相催化反应装置购于天津天大北洋公司。
本发明实施例中采用的主要试剂:八水氧氯化锆、氢氧化钠、氨水、氯金酸均购置于国药集团化学试剂有限公司;纯氢、纯氮和高纯一氧化碳均购置于康龙医用气体有限公司。
各指标的表征:
(1)x-射线粉末衍射(xrd):催化剂或载体的体相机构、晶相组成及微观结构用荷兰panalytic公司的x'pertpro衍射仪上进行,采用x'celerator探测器,cu-kα(λ=0.1541nm)靶辐射,管电压40kv,管电流40ma,扫描步长0.12°/min,扫描范围为10°到140°。(2)拉曼光谱表征(raman):仪器为renishawinviaplus,在室温下使用半导体激光器作为照射源(波长=532nm)收集拉曼光谱。
(3)x-射线光电子能谱(xps):在vgescalab250型(美国热电公司)光电子能谱仪上进行;所有电子结合能(be)以c1s(284.5ev)为内标进行校准。
(4)比表面积表征(bet):在micrometricsasap2020仪器上进行;通过使用氮气作为吸附气体,测量样品比表面积,孔体积和孔径分布。
(5)高分辨透射电镜分析(hrtem):使用jeol2010f(200kv)型场发射高分辨率透射电子显微镜观察样品的形貌。催化剂样品研磨后溶于乙醇,经超声震荡20min后,取上层悬浮液滴置于镀碳膜铜网,自然晾干后在200kv电子束下观察粒子形貌和结晶程度。
(6)场发射扫描电子显微镜(sem):使用hitachis-4800场发射扫描电镜观察形貌。
(7)电子顺磁共振(epr):在室温下,使用brukeremx-8光谱仪在9.44ghz下收集电子顺磁共振(epr)光谱。
(8)荧光光谱仪(pl):使用hitachif-4500荧光光谱仪测试,激发光源为300nm。
(9)莫特-肖特基曲线(mott-schottkyplots):使用chi660d电化学工作站,在黑暗中以10khz的频率进行电化学阻抗测量。电解液为1m的氢氧化钠溶液(ph为13.6),饱和甘汞电极和铂电极分别为参比电极和对电极。
实施例1改性zro2载体(zro2-ht-h)和改性au-zro2催化剂(au-zro2-ht-h)的制备
一、改性zro2载体的制备
具体制备过程如下:
(1)采用水热法(ht)制备zro2载体:取60ml、0.40mol/l的zrocl2·8h2o水溶液装入100ml聚四氟乙烯罐子中,将其放入不锈钢高压反应釜中,旋紧,在150℃水热反应6小时;然后,对水热产物进行高速离心洗涤数次,直到上层清液的ph值为中性,所得沉淀于70℃烘干过夜,制得zro2载体。
(2)将zro2载体置于氢气(h)气氛下550℃焙烧4小时,得到改性的zro2载体(简写为:zro2-ht-h)。
二、改性au-zro2催化剂
采用并流共沉淀法,利用上述改性zro2载体(zro2-ht-h),分别制备含4wt.%au的au-zro2催化剂(au-zro2-ht-h)。
具体方法如下:
(1)取0.7g上述改性zro2载体,加入到200ml去离子水中,调节ph为8±1,将其作为底液;
(2)在取27wt.%的氨水0.4ml加入100ml去离子水中,记为溶液c;再取60ml0.0025mol/lhaucl4溶液,记为溶液d;
(3)同时将c和d溶液通过蠕动泵逐滴加入到底液中,强烈搅拌,保持反应温度在80℃,ph为8±1;
(4)滴加完成后,继续在80℃下老化6小时;将所得沉淀物经过抽滤、洗涤数次脱除杂质离子,再经70℃干燥过夜,制备得到改性au-zro2催化剂。
对比例1改性zro2载体(zro2-ht-a)和改性au-zro2催化剂(au-zro2-ht-a)的制备
一、改性zro2载体的制备
具体制备过程如下:
(1)采用水热法(ht)制备zro2载体:取60ml、0.40mol/l的zrocl2·8h2o水溶液装入100ml聚四氟乙烯罐子中,将其放入不锈钢高压反应釜中,旋紧,在150℃水热反应6小时;然后,对水热产物进行高速离心洗涤数次,直到上层清液的ph值为中性,所得沉淀于70℃烘干过夜,制得zro2载体。
(2)将zro2载体置于静态空气(a)气氛下550℃焙烧4小时,得到改性的zro2载体(简写为:zro2-ht-a)。
二、改性au-zro2催化剂
具体制备方法同实施例1。
对比例2改性zro2载体(zro2-ht-a-h)和改性au-zro2催化剂(au-zro2-ht-a-h)的制备
一、改性zro2载体的制备
具体制备过程如下:
(1)采用水热法(ht)制备zro2载体:取60ml、0.40mol/l的zrocl2·8h2o水溶液装入100ml聚四氟乙烯罐子中,将其放入不锈钢高压反应釜中,旋紧,在150℃水热反应6小时;然后,对水热产物进行高速离心洗涤数次,直到上层清液的ph值为中性,所得沉淀于70℃烘干过夜,制得zro2载体。
(2)将zro2载体置于静态空气(a)气氛下550℃焙烧4小时,再在氢气(h)气氛下550℃焙烧4小时,得到改性zro2载体(简写为:zro2-ht-a-h)。
二、改性au-zro2催化剂
具体制备方法同实施例1。
对比例3改性zro2载体(zro2-p-h)和改性au-zro2催化剂(au-zro2-p-h)的制备
一、改性zro2载体的制备
具体制备过程如下:
(1)采用沉淀法(p)制备二氧化锆:称取4.51g的zrocl2·8h2o(ar,国药集团化学试剂有限公司)置于70ml水中,搅拌均匀。记为a溶液。配置0.5mol/l的naoh溶液200ml,记为b溶液。将a和b溶液通过蠕动泵同时逐滴加入到200ml蒸馏水中,强烈搅拌,保持反应温度在65℃,ph为10±1.滴加完成后,继续老化2小时。然后将所得产物经过多次沉降,倒掉上层清液,直到上层清液ph为中性,在70℃烘干过夜,制得zro2载体。
(2)将上述zro2载体置于氢气(h)气氛下550℃焙烧4小时,得到改性的zro2载体(简写为:zro2-p-h)。
二、改性au-zro2催化剂
具体制备方法同实施例1。
对比例4改性zro2载体(zro2-p-a)和改性au-zro2催化剂(au-zro2-p-a)的制备
一、改性zro2载体的制备
具体制备过程如下:
(1)采用沉淀法(p)制备二氧化锆:称取4.51g的zrocl2·8h2o(ar,国药集团化学试剂有限公司)置于70ml水中,搅拌均匀。记为a溶液。配置0.5mol/l的naoh溶液200ml,记为b溶液。将a和b溶液通过蠕动泵同时逐滴加入到200ml蒸馏水中,强烈搅拌,保持反应温度在65℃,ph为10±1.滴加完成后,继续老化2小时。然后将所得产物经过多次沉降,倒掉上层清液,直到上层清液ph为中性,在70℃烘干过夜,制得zro2载体。
(2)将zro2载体置于静态空气(a)气氛下550℃焙烧4小时,得到改性的zro2载体(简写为:zro2-p-a)。
二、改性au-zro2催化剂
具体制备方法同实施例1。
对比例5改性zro2载体(zro2-p-a-h)和改性au-zro2催化剂(au-zro2-p-a-h)的制备
一、改性zro2载体的制备
具体制备过程如下:
(1)采用沉淀法(p)制备二氧化锆:称取4.51g的zrocl2·8h2o(ar,国药集团化学试剂有限公司)置于70ml水中,搅拌均匀。记为a溶液。配置0.5mol/l的naoh溶液200ml,记为b溶液。将a和b溶液通过蠕动泵同时逐滴加入到200ml蒸馏水中,强烈搅拌,保持反应温度在65℃,ph为10±1.滴加完成后,继续老化2小时。然后将所得产物经过多次沉降,倒掉上层清液,直到上层清液ph为中性,在70℃烘干过夜,制得zro2载体。
(2)将zro2载体置于静态空气(a)气氛下550℃焙烧4小时,再在氢气(h)气氛下550℃焙烧4小时,得到改性zro2载体(简写为:zro2-p-a-h)。
二、改性au-zro2催化剂
具体制备方法同实施例1。
对比例6
将对比例1的常规au-zro2-ht-a催化剂在氢气气氛下300℃焙烧4小时,并在氢气气氛下降至室温取出,获得的催化剂简写为au-zro2-ht-a-300。
对比例7
将对比例1的常规au-zro2-ht-a催化剂在氢气气氛下550℃焙烧4小时,并在氢气气氛下降至室温取出,获得的催化剂简写为au-zro2-ht-a-550。
检测上述实施例1和对比例1-7制备获得的催化剂,对其进行催化活性分析、sem分析、bet比表面和孔结构分析、xps分析以及光电性能分析。
1、催化活性分析
如图1a所示,以氢气气氛下焙烧制备的zro2-ht-h为载体的au-zro2-ht-h催化剂表现出较高的催化活性,其在200℃和300℃下的co转化率分别达到了68.6%和92.5%。对比发现,各种催化剂的活性顺序为:au-zro2-ht-h>au-zro2-ht-a-h>au-zro2-ht-a。具体来说,相比对比例1和2,au-zro2-ht-h催化剂在200℃的co转化率分别提升了55%(从44.2%提升到68.6%)和28%(从53.4%提升到68.6%)。可见,实施例1的改性zro2-ht-h载体能够促使催化剂表现出较高的催化活性。也就是说,对于水热法制备的zro2载体,氢气焙烧的改性方法能够有效显提高催化剂的催化活性。
类似地,从图1b可见,各种催化剂的活性顺序为:au-zro2-p-h>au-zro2-p-a-h>au-zro2-p-a。具体来说,与au-zro2-p-a-h和au-zro2-p-a相比,au-zro2-p-h催化剂在200℃的co转化率分别提升了90%(从27.9%提升到53%)和16%(从45.7%提升到53%)。可见,对于沉淀法,改性zro2-p-h载体能够促使催化剂表现出较高的催化活性。也就是说,对于水沉淀法制备的zro2载体,氢气焙烧的改性方法同样能够有效显提高催化剂的催化活性。
此外,为了讨论氢气气氛焙烧对载体和催化剂的影响,将对au-zro2-ht-a催化剂分别在300℃和550℃进行氢气气氛下的焙烧,即获得了au-zro2-ht-a-300(对比例6)和au-zro2-ht-a-550(对比例7)催化剂。对比发现,au-zro2-ht-a-300和au-zro2-ht-a-550的催化活性均明显低于au-zro2-ht-a,这可能是由于负载活性金之后的催化剂,再经受300℃和550℃的高温焙烧后,金纳米颗粒产生团聚现象,导致其催化剂活性不佳。
所以,氢气焙烧的方法只适用于载体改性,并不适用于催化剂改性。另外,au-zro2-ht-a-550的催化活性高于au-zro2-ht-a-300,这可能是由于,虽然550℃的高温可能比300℃引起更严重的金纳米颗粒团聚,但是其催化剂载体可以在550℃氢气焙烧中得到改性,故表现出略高的活性,而300℃氢气焙烧温度不足以改性载体。因此,氢气焙烧的改性方法需要在较高的温度下进行。综上所述,对于载体进行氢气高温焙烧改性,负载活性金后,其催化剂的活性明显得到提升。
2、xrd分析
如图2所示,经550℃不同气氛下焙烧后,载体zro2在24°、24.4°、28.1°、31.4°、34.1°、34.3°和35.3°等位置出现了单斜相的特征峰,这与zro2数据库中峰的位置一致(jcpds01-089-9066),没有杂相出现。经过金负载后的au-zro2催化剂的xrd衍射峰与载体几乎一样,说明负载后载体的晶相得到了很好的保持,550℃焙烧后的二氧化锆的晶粒尺寸并没有发生变化,也没有发现金属au的衍射峰,表明au高度分散在载体上。
3、sem分析
如图3所示,由实施例1和对比例1制备的改性zro2载体中的zro2颗粒表现出规则的圆形片状结构形貌。
4、tem分析
如图4中的a,b和g所示,在au-zro2-ht-h,au-zro2-ht-a-h和au-zro2-ht-a催化剂上,au颗粒的分散程度和尺寸大小几乎都相同,表明它们的催化活性之间的差异与au无关,那么只能说明与zro2载体的差异有关。
如图4c所示,zro2-ht-h纳米晶体表面出现一层晶格条纹不明显的区域(无序层),厚度为约2nm。然而,在zro2-ht-a-h和zro2-ht-a的hrtem图像上,并没有发现在表面区域附近有明显的无序层(图2-4e,h)。
如图4f和i所示,当au负载在zro2载体上之后,尽管表面无序层减小到约1nm厚,但au-zro2-ht-h依旧保留了由结晶核和无序层组成的微结构。同比,au-zro2-ht-a-h和au-zro2-ht-a催化剂上仍然没有明显的无序层。
结果表明,h2气氛导致zro2载体表面结构重排。同时为了验证其稳定性,我们将氢气焙烧后的载体置于空气中暴露几个月,并且在制备au催化剂的过程中长时间浸入水中,结果发现,其表面的无序层并没有被破坏,证明其有很高的稳定性。
5、bet比表面和孔结构分析
从表1中可以看出,与zro2-ht-a和zro2-ht-a-h相比,zro2-ht-h比表面(24.6m2/g)最大。可见,氢气焙烧的改性方法可明显提升载体的比表面积。
表1zro2载体和au/zro2催化剂织构性能表
如图5所示,实施例1和对比例1、2制备的载体具有2个滞后环,属于双介孔型zro2。从图中可以看出zro2-ht-h载体的滞后环中吸附、脱附曲线分离处的相对压力(p/p0)最小。根据kelvin方程,孔径越小,凝聚所需的饱和蒸气压越低,滞后环中吸附和脱附曲线分离处的相对压力越低,说明毛细孔的孔径越小,反之亦然。说明zro2-ht-h载体的孔径最小,这与表1中zro2-ht-h载体具有最小的平均孔径(30nm)的结果一致。
如图6所示,载体和催化剂的孔径主要分布两种,分别为3~6nm的介孔和30~40nm左右的堆砌孔,且负载金后,孔径分布并未改变。其中,zro2-ht-h载体的介孔孔径最小,而zro2-ht-a载体和zro2-ht-a-h载体的介孔孔径几乎一样。可见,氢气焙烧的改性方法导致了介孔孔径明显变小。这是因为氢气焙烧过程中,圆形片状结构的zro2载体的表面具有无序层,形成较多的氢刻蚀小孔。关联催化剂活性,发现载体的孔径变化与对应的催化剂的活性的变化基本一致,孔径越小,催化剂的活性越好。
根据表1中的比表面大小的比较,zro2-ht-h载体不仅孔径小,且比表面大,说明其内部具有很多比zro2-ht-a载体和zro2-ht-a-h载体更多更小的微孔结构,提高了孔内表面的利用,从而表现出更好的催化活性。
6、xps表征
如图7a所示,zro2-ht-h和zro2-ht-a-h都具有比zro2-ht-a少得多的表面-oh。说明zro2-ht-h和zro2-ht-a-h中存在的氧原子量比常态下的二氧化锆少,且zro2-ht-h显示出了更少的表面-oh。结果表明,在氢气焙烧过程中,由于h2还原导致表面-oh的减少,从而形成表面氧空位。
如图7b所示,au-zro2-ht-h中仅仅出现了au0物质(价态为0价的单质金),而au-zro2-ht-a和au-zro2-ht-a-h中不仅出现了au0,同时还出现了au3+。考虑到本发明采用的金源是haucl4(au3+),所以推断au0是由于载体表面被还原的氧空位上的电荷转移到au颗粒所得到的。因此,由于在zro2-ht-h上的表面氧空位远远大于zro2-ht-a和zro2-ht-a-h上的表面氧空位,所以在金(au(oh)3)沉积期间,负载在zro2-ht-h上的au0的量远比在zro2-ht-a和zro2-ht-a-h上的多。而对于co氧化反应,相比于au3+,au0在au-zro2催化剂中起到更好的催化效果,所以au-zro2-ht-h表现出更高的催化活性。
结果表明,催化剂的催化活性好坏不取决于它们的金负载量的差异,而是取决于au价态的差异,而这受到载体表面氧空位数量的影响。
7、epr表征
载体和催化剂的表面氧空位可进一步通过epr表征结果进行证实。如图8所示,zro2-ht-h和au-zro2-ht-h均产生明显的epr信号,这应归因于吸附在氧空位处的o2(来自空气)捕获的单电子o2·-自由基,然而,在zro2-ht-a,au-zro2-ht-a,zro2-ht-a-h和au-zro2-ht-a-h中却没有发现信号。结果表明,氢气焙烧的改性方法导致在zro2载体上产生更多的表面氧空位。
8、pl表征
图9是各种zro2载体和au-zro2催化剂的荧光光谱图。从图中可以看出,相比zro2-ht-a载体,zro2-ht-a-h和zro2-ht-h载体中的电子不易从激发态跃迁回基态,其电子-空穴复合机率降低。同样,在负载au之后,au-zro2-ht-a-h和au-zro2-ht-h催化剂的荧光强度也比au-zro2-ht-a催化剂弱,尤其是au-zro2-ht-h表现出极低的荧光强度,其电子-空穴复合机率同样降低。因此,氢气焙烧的改性方法可有效降低载体和催化剂中电子-空穴复合机率。
9、光电性能分析
在黑暗中进行各种zro2载体和au-zro2催化剂的电化学阻抗测量,频率为10khz,以研究氢气焙烧对其电子性能的影响。如对于n型半导体所预期的,所有样品在mott-schottky图中都显示为正斜率(图10)。这些样品的供体密度由mott-schottky图中的斜率计算:
nd=(2/e0εε0)[d(1/c2)/dv]-1
其中e0是电子电荷,ε是tio2的介电常数(ε=170),ε0是真空介电常数(8.85e-12f/m),nd是供体密度,v是电极处的施加偏压。
zro2-ht-a,zro2-ht-a-h和zro2-ht-h计算得到的电子密度分别为2.5×1022cm-3,9.6×1022cm-3和3.4×1023cm-3。同时,在负载au之后,电子密度大小顺序没有发生改变,依旧为au-zro2-ht-h(4.3×1024cm-3)>au-zro2-ht-a-h(7×1023cm-3)>au-zro2-ht-a(1.7×1023cm-3),且相比对应的载体,电子密度增加了约10倍。由于在氢气焙烧后样品形态没有发生明显变化(如图3所示),说明氢气焙烧的改性方法可有效增加载体的供体密度。增加的供体密度是由于氧空位的增加,因为氧空位是二氧化锆的电子供体。增加的供体密度改善了zro2中的电荷传输,降低了电子-空穴复合机率,从而提升了催化剂的催化活性。
- 还没有人留言评论。精彩留言会获得点赞!