改性无机耐热氧化物及其制备方法及加氢脱硫催化剂和降低加氢脱硫催化剂失活速度的方法与流程
本发明是关于一种改性无机耐热氧化物及其制备方法和应用以及使用该改性无机耐热氧化物作为载体的加氢脱硫催化剂和降低加氢脱硫催化剂失活速度的方法。
背景技术:
加氢处理是现代炼油工业中的支柱技术,在生产清洁燃料、提高产品质量、充分利用石油资源和原料预处理等方面发挥着重要作用。随着经济、环保和社会的发展,使得炼油企业对加氢处理催化剂的活性和稳定性不断提出更高的要求。常见加氢处理催化剂以VIII族金属Co、Ni作助活性组元,以VIB族金属Mo、W作主活性组元,载体常选用氧化铝。
例如,CN201010236483.7公开了一种以氧化硅-氧化铝为载体的加氢脱硫催化剂,含有氧化钴2~6wt%,氧化钼9~15wt%,碱土金属氧化物2~8wt%,氧化磷2~6wt%,碱金属氧化物3~5wt%,氧化硅2~6wt%,氧化铝54~80wt%。催化剂的加氢活性和选择性高,稳定性好。
CN201010269179.2描述了一种加氢脱硫催化剂及其制备方法,所述催化剂是以钼、镍、锌、铜复合金属氧化物至少两种为活性组分,添加Si、Ti、Ca、Ce、Mg、P氧化物至少一种为助剂,余量为氧化铝。
CN200710177578.4涉及一种组合氧化铝基的选择性加氢脱硫催化剂及其制备方法,该方法将大孔氧化铝和小孔氧化铝复合,然后依次负载助剂镁和硼以及活性组分钴和钼,再经干燥和焙烧,制成催化剂。
CN200710177577.X公开了一种催化剂以氧化铝和氧化硼为载体,并经 过多元助剂镁、钾和磷修饰,担载活性组分钴和钼。
CN200480005056.0公开了一种含有非贵金属的第VIII族金属、一种非贵金属的第VIB族金属和一或多种选自铝、硅、镁、钛、锆、硼和锌的元素的催化剂组合物。
CN201210268290.9公开了载体由氧化铝、氧化硅、氧化镁组成、而所述活性组分由WO3和NiO或MoO3和NiO组成、并含有以K2O和Ga2O3组成的助剂的催化剂。
CN96120875.9公开了含有活性组分Ni、W、Co,助催化剂成分为P或P和Mg的混合物,载体为γ-Al2O3。
CN96106586.9公开了一种催化剂,它由γ-Al2O3负载Ni、W、Co活性组分,还可以加入选自Mg、Zn、Fe、Ca中的任一元素作为助催化剂成分。
尽管现有技术对催化剂的组成及其制备方法进行了大量的研究,催化剂的性能也取得了较大的进步。然而,随着社会对燃料清洁程度要求的增加以及原油劣质化加剧的趋势,加氢处理催化剂的性能需要进一步提高。
技术实现要素:
为了克服现有技术的不足,本发明提供了一种能够制备性能更为优越的催化剂的改性无机耐热氧化物及其制备方法和应用及加氢脱硫催化剂。
文献报道,在氧化铝的表面存在多种具有不同强度的酸性羟基和碱性羟基(C.Morterra et al.,Catalysis Today 27(1996)497-532)。在氧化态催化剂中,主活性组分(如Mo或W)以氧化物的形式分散在载体的表面,Mo或W物种与氧化铝表面的各种羟基作用形成化学键。形成的化学键在硫化过程中发生断裂并释放出金属,释放的金属与硫发生反应生成硫化态活性相。硫化态活性相的分散性质对催化剂的活性具有重要影响。本发明的发明人发现,通过优化载体表面羟基的性质,能够使活性组分更好的分散,从而获得更多活 性中心,由此在实际应用过程中催化剂活性更高,并且催化剂的失活速率得到降低,可以延长催化剂的使用寿命。
基于此,根据本发明的第一方面,本发明提供了一种改性无机耐热氧化物,该改性无机耐热氧化物具有酸性羟基和碱性羟基,红外光谱中,强酸性羟基峰在3640-3700cm-1附近,强碱性羟基峰在3740-3800cm-1附近,其特征在于,所述强碱性羟基峰的相对高度为30-90%,所述强碱性羟基峰与所述强酸性羟基峰的峰高比为1:0.85-1.15。
根据本发明的第二方面,本发明提供了一种改性无机耐热氧化物的制备方法,该方法包括将无机耐热氧化物和/或无机耐热氧化物的前身物、助挤剂、胶溶剂混合均匀后进行成型,然后将所得成型物依次进行干燥和焙烧,其特征在于,该方法还包括在成型前和/或成型后加入酸性羟基中和剂和碱性羟基中和剂,所述酸性羟基中和剂为碱土金属氧化物、碱土金属硝酸盐和碱土金属氢氧化物中的一种或多种,所述碱性羟基中和剂为含硼、镓、硅、锗、磷、氟元素中的一种或多种的氧化物、含氧酸中的一种或多种,所述酸性羟基中和剂和碱性羟基中和剂的用量使得所得耐热无机氧化物的红外光谱中,强碱性羟基峰的相对高度为30-90%,强酸性羟基峰的相对高度为20-90%,所述强酸性羟基峰在3640-3700cm-1附近,强碱性羟基峰在3740-3800cm-1附近。
根据本发明的第三方面,本发明提供了一种由上述制备方法制得的改性无机耐热氧化物。
根据本发明的第四方面,本发明提供了一种由上述改性无机耐热氧化物作为催化剂载体的应用。
根据本发明的第五方面,本发明提供了一种加氢脱硫催化剂,该加氢脱硫催化剂含有载体和负载在载体上的金属活性成分,其特征在于,所述载体为本发明上述提供的改性无机耐热氧化物。
根据本发明的第六方面,本发明提供了一种降低加氢脱硫催化剂失活速 度的方法,该方法包括采用上述方法制备改性无机耐热氧化物,然后以该改性无机耐热氧化物为载体负载具有加氢脱硫活性的金属活性成分。
本发明提供的改性无机耐热氧化物由于强碱性羟基峰与强酸性羟基峰的峰高相当,使得其作为载体时的催化剂活性更高,并且催化剂的失活速率得到降低,可以延长催化剂的使用寿命。本发明提供的加氢脱硫催化剂通过使用上述改性无机耐热氧化物作为载体并进一步配合特定的金属活性组分含量,从而催化剂的活性进一步明显提高,催化剂的失活速率进一步明显降低。
本发明的其它特征和优点将在随后的具体实施方式部分予以详细说明。
附图说明
附图是用来提供对本发明的进一步理解,并且构成说明书的一部分,与下面的具体实施方式一起用于解释本发明,但并不构成对本发明的限制。在附图中:
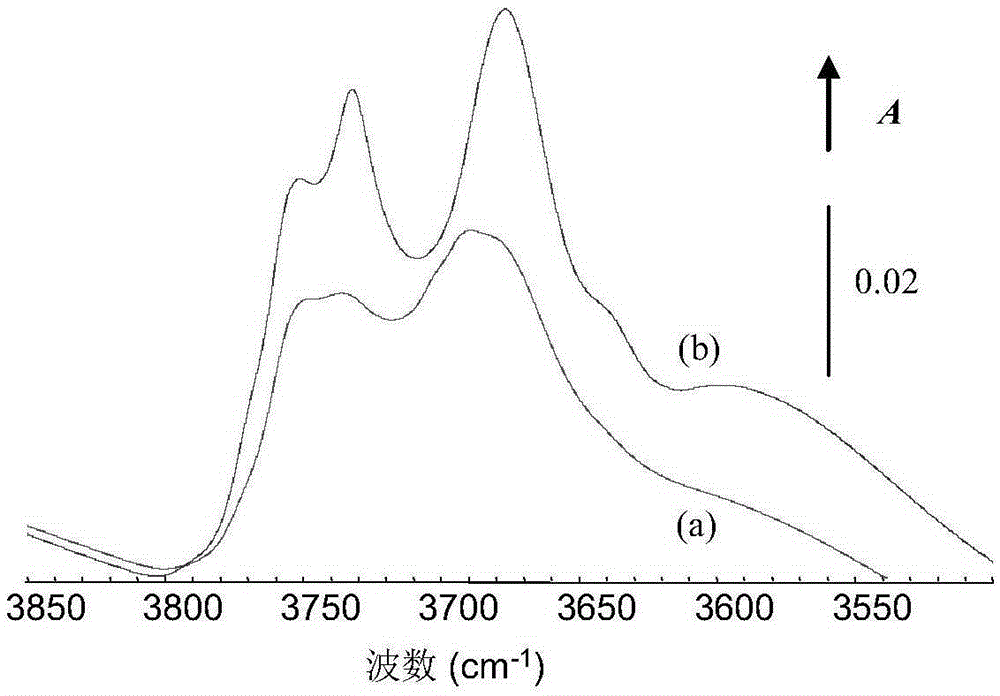
图1中(a)是实施例1制得的氧化铝的红外光谱曲线,(b)是对比例1制得的氧化铝的红外光谱图。
具体实施方式
以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
根据本发明的第一方面,本发明提供了一种改性无机耐热氧化物,该改性无机耐热氧化物具有酸性羟基和碱性羟基,红外光谱中,强酸性羟基峰在3640-3700cm-1附近,强碱性羟基峰在3740-3800cm-1附近,其特征在于,所述强碱性羟基峰与所述强酸性羟基峰的峰高比为1:0.85-1.15,优选为1:0.9-1.1。
本发明中,所述强碱性羟基峰与所述强酸性羟基峰的峰高相当,由此所述强碱性羟基与所述强酸性羟基数量相当,使得其作为载体时的催化剂活性更高,并且催化剂的失活速率得到降低,可以延长催化剂的使用寿命。而现有技术中,无机耐热氧化物的所述强碱性羟基峰与所述强酸性羟基峰的峰高比通常为1:1.2-1.4。
优选情况下,本发明提供的无机耐热氧化物的红外谱图中,所述强碱性羟基峰的相对高度为30-90%,优选为40-85%,更优选为55-75%;所述强酸性羟基峰的相对高度为20-90%,优选为50-85%,更优选为60-80%。
本发明中,羟基峰的相对高度的含义与现有相对结晶度的含义类似,具体表示本发明的改性无机耐热氧化物的羟基峰的高度与参比无机耐热氧化物的相应羟基峰的高度比,即,相对于参比无机耐热氧化物,碱性羟基峰高度降低幅度控制为10-70%,优选为15-60%;强酸性羟基峰的降低幅度控制为10-80%,优选为15-40%。所谓参比无机耐热氧化物是指制备过程中不引入碱性羟基中和剂和酸性羟基中和剂时制备的无机耐热氧化物,即常规的无机耐热氧化物。
本发明中,两种酸性羟基变化幅度可以相同,也可以不同,只要最终的高度比或相对高度控制在上述范围内即可。由于氧化铝表面中还存在其他类型羟基,如弱酸性和弱碱性羟基,在调变过程中这些羟基的数量可能发生变化,其变化量不作为控制指标。另外新的羟基也可能生产,属于正常现象,不做限制。
本发明中,所述红外光谱图是首先采用自支撑压片法获得样品,然后在红外池中对样品进行预处理后再用红外光谱仪扫描得到样品的羟基谱图(以下称为自支撑压片法)。自支撑压片法为本领域熟知技术,不再赘述。
在进行载体羟基测量前,对样品进行预处理,预处理在400℃下进行3h以上,处理后将温度降至室温。该测量方法为本领域中常用技术,在此不再 详述。
根据本发明提供的改性无机耐热氧化物可以是现有技术各种能够用作催化剂载体的无机耐热氧化物的改性物,例如,可以为氧化铝、氧化硅、氧化锆、氧化铝-氧化钛和氧化铝-氧化硅中的一种或多种的改性物。所谓的改性物即为强碱性羟基与弱碱性羟基数量相当从而强碱性羟基峰与所述强酸性羟基峰的峰高比为1:0.85-1.15,优选为1:0.9-1.1并且进一步优选碱性羟基和酸性羟基均有所弱化的产物。
所述无机耐热氧化物改性前后的形状均可以是现有技术已知的各种使用用作催化剂载体的形状,例如可以为颗粒状、条状或三叶草状,且无机耐热氧化物的大小为0.2-15毫米。
本发明中,(改性)无机耐热氧化物的大小是指无机耐热氧化物从一点到另一点的最大直线距离,可以通过筛分获得。
根据本发明的第二方面,本发明提供了一种无机耐热氧化物的制备方法,该方法包括将无机耐热氧化物和/或无机耐热氧化物的前身物、助挤剂、胶溶剂混合均匀后进行成型,然后将所得成型物依次进行干燥和焙烧,其特征在于,该方法还包括在成型前和/或成型后加入酸性羟基中和剂和碱性羟基中和剂,所述酸性羟基中和剂为碱土金属氧化物、碱土金属硝酸盐和碱土金属氢氧化物中的一种或多种,所述碱性羟基中和剂为含硼、镓、硅、锗、磷、氟元素中的一种或多种的氧化物、含氧酸中的一种或多种,所述酸性羟基中和剂和碱性羟基中和剂的用量使得所得耐热无机氧化物的强碱性羟基峰的相对高度为30-90%,强酸性羟基峰的相对高度为20-90%,所述强酸性羟基峰在3640-3700cm-1附近,强碱性羟基峰在3740-3800cm-1附近。
需要说明的是,本发明的中和并非通常意义上的酸碱中和,而是改变羟基性质的中和,具体的是降低羟基峰高度的反应。例如,尽管硝酸具有酸性,能够中和碱性物质,但并不能用作本发明的碱性羟基中和剂。实验证明,无 论是加大作为胶溶剂时的硝酸的用量,还是在后续混合后或成型过程中额外加入硝酸,均不能起到降低羟基峰高度的作用。
根据本发明,优选情况下,所述酸性羟基中和剂为氢氧化铍、氢氧化镁、氢氧化钙、氢氧化钡、氧化铍、氧化镁、氧化钙、氧化锶、氧化钡、硝酸钡、硝酸钙、硝酸镁、硝酸铍中的一种或多种,进一步优选为氢氧化铍、硝酸钙和硝酸镁中的一种或多种。
根据本发明,优选情况下,所述碱性羟基中和剂为HF、氟硅酸、硅溶胶、硼酸、磷酸、亚磷酸、锗酸中的一种或多种,进一步优选为HF、氟硅酸、硅溶胶、硼酸中的一种或多种。
根据本发明提供的改性无机耐热氧化物的制备方法,相对于1摩尔的无机耐热氧化物的前身物,所述酸性羟基中和剂的用量为0.02-0.08摩尔,所述碱性羟基中和剂的用量为0.02-0.1摩尔。本发明的发明人发现,通过将中和剂的用量控制在上述范围内,基本可以使碱性羟基下降幅度在前述值范围内(30-90%),使酸性羟基下降幅度在前述值范围内(20-90%)。由于不同的无机耐热氧化物中羟基量不同,可以通过进一步实验确定使所述强碱性羟基峰与所述强酸性羟基峰的峰高比为1:0.85-1.15,优选为1:0.9-1.1范围内的具体的用量。具体可以参照以下方法来确定:
按照常规方法制备纯氧化铝载体,然后采用孔饱和浸渍法或混捏法制备添加元素的摩尔分数为5%的载体,利用红外光谱法测量强碱性或强酸性羟基的相对高度。利用两点直线法确定相对高度在所需范围内时元素的用量。例如,氧化铝中引入摩尔分数为5%的硼,对应强碱性羟基相对高度为53%。由于强碱性羟基相对高度与元素含量成反比例关系,因此可以计算出强碱性羟基相对高度为30-90%时对应的硼元素的摩尔分数。同理,其它元素相应的羟基相对高度范围的摩尔分数范围也可以实际测出。
需要特别强调的是,必须严格控制羟基中和剂的用量大于上述范围的下 限值且小于上述范围的上限值,才能获得降低羟基峰高度且降低幅度在上述范围内的效果。事实上,现有技术也在载体的制备过程中加入B、P和Mg作为助剂成分,但它们或者只加入其中同一种性质如仅加入酸性中和剂或者仅加入碱性中和剂;或者加入的量较大或者较小,因而不能达到同时降低酸性羟基和碱性羟基峰高度的效果,因此催化剂的加氢脱硫性能也就不能获得明显提升。
根据本发明提供的改性无机耐热氧化物的制备方法,所述酸性羟基中和剂和碱性羟基中和剂可以同时加入,也可以各自单独且不同时加入,可以先加酸性羟基中和剂,后加碱性羟基中和剂,也可以先加碱性羟基中和剂,后加酸性羟基中和剂。它们可以在制备过程中的各个步骤加入,例如,可以与无机耐热氧化物的前身物、助挤剂和胶溶剂一起混合均匀后进行成型;也可以在无机耐热氧化物的前身物、助挤剂和胶溶剂一起混合均匀后再先后加入,然后进行成型;还可以在无机耐热氧化物的前身物、助挤剂和胶溶剂一起混合均匀后加入酸性羟基中和剂或碱性羟基中和剂,成型、干燥并焙烧后再加入碱性羟基中和剂或酸性羟基中和剂。成型后再加入的方式优选为孔饱和浸渍法。如果在焙烧成型后再加入碱性羟基中和剂或酸性羟基中和剂,则需要对载体再次进行焙烧。并且第二次焙烧的温度要低于前一次焙烧温度,例如低50-200℃,其余条件保持相同。
本发明的制备方法中,成型的方式可以采用现有的各种催化剂载体成型方法,例如可以是挤条成型法。
成型后可以直接焙烧,也可以先干燥后焙烧,优选为先干燥后焙烧。干燥的温度可以为60-150℃,优选为100-140℃;干燥的时间可以为1-10小时,优选为3-6小时。焙烧的温度可以为300-800℃,优选为400-700℃;焙烧的时间可以为2-10小时,优选为3-5小时。
根据本发明提供的改性无机耐热氧化物的制备方法,所述无机耐热氧化 物和/或其前身物可以根据所需的改性无机耐热氧化物进行适当的选择,具体的,所述无机耐热氧化物和/或其前身物可以为拟薄水铝石、薄水铝石、无定型氢氧化铝、三水铝石、氧化硅、氧化锆、氧化铝-氧化钛和氧化铝-氧化硅中的一种或多种。
所述助挤剂和胶溶剂均可以参照现有技术进行选择,例如,助挤剂可以为田菁粉、甲基纤维素和淀粉等物质中的一种或多种,胶溶剂可以为硝酸、柠檬酸和醋酸中的一种或多种。
本发明第三方面还提供了由上述制备方法制得的改性无机耐热氧化物。
本发明第四方面还提供了改性无机耐热氧化物作为催化剂载体的应用。
采用本发明提供的上述改性无机耐热氧化物作为催化剂载体,能够有效提高催化剂的加氢脱硫活性。
为此,本发明第五方面还提供了一种加氢脱硫催化剂,该加氢脱硫催化剂含有载体和负载在载体上的金属活性成分,其特征在于,所述载体为本发明上述提供的改性无机耐热氧化物。
本发明第六方面还提供了一种降低加氢脱硫催化剂失活速度的方法,该方法包括采用上述方法制备改性无机耐热氧化物,然后以该改性无机耐热氧化物为载体负载具有加氢脱硫活性的金属活性成分。
所述加氢脱硫催化剂的金属活性成分可以为各种具有加氢脱硫活性的金属组分,例如可以为元素周期表中VIII族和VIB族金属元素,优选地,所述金属活性成分为镍和/或钴以及钨和/或钼。
所述加氢脱硫催化剂中,载体表面VIB族金属元素的原子浓度为2-8个原子/nm2,优选为2.5-6个原子/nm2;VIII/(VIII+VIB)原子比范围是0.2-0.4,优选为0.3-0.4。当所述金属活性成分为镍和/或钴以及钨和/或钼时,载体表面Mo和/或W的总原子浓度为2-8个原子/nm2,优选为2.5-6个原子/nm2;Co(Ni)/(Co(Ni)+Mo+W)原子比范围是0.2-0.4,优选为0.3-0.4。
本发明中,载体表面金属原子浓度由金属的原子总数与载体比表面积之比计算而得,载体的比表面积由常规氮气吸附法测得。
上述加氢脱硫催化剂可以通过将加氢活性金属组分负载在上述改性无机耐热氧化物上来制得。例如可以通过孔饱和浸渍法。具体可以通过下述两种方式来负载。方式1:以硝酸钴、硝酸镍、七钼酸铵和偏钨酸铵为原料,在添加或者不添加助溶剂的情况下配制成浸渍溶液。助溶剂主要包括氨水,含氮有机化合物,如乙二胺、EDTA、氨基三乙酸、环己二胺四乙酸和氨基酸类。这些物质与Co(Ni)的摩尔比为0.5-2。方式2:以碱式碳酸钴、碱式碳酸镍、氧化钼、偏钨酸铵和磷酸为原料,或添加一种和/或多种含有羟基和/或羧基的有机化合物为助溶剂配制浸渍液。优选选用酒石酸、柠檬酸、乙二醇、丙三醇等。磷与Co(Ni)的原子比为0.5-2,有机物与Co(Ni)的原子比为0.5-2。以上述方法配制的浸渍液浸渍载体0.5-4h之后,在50-180℃下干燥1-5h,然后在300-500℃下焙烧2-5h制备成催化剂。
本发明提供的加氢脱硫催化剂由于其中的强碱性和强酸性羟基数量降低,一方面使得活性组分Mo或W与羟基的作用力降低提高了其可硫化能力,另一方面羟基数量的减少使得结焦物质在催化剂上的吸附性能降低进而降低了催化剂的失活速率,可以延长运转周期。同时,由于强碱性和强酸性羟基数量的减少,使得载体表面的羟基酸碱性趋向更加均匀,进而使活性组分更为均匀的分散。由此方法制备催化剂的加氢脱硫活性以及活性稳定性更为优越。
下面的实施例将对本发明做进一步的说明,但是这些实施例并不能限制本发明。以下实施例中使用的氢氧化铝粉为长岭催化剂厂生产的拟薄水铝石。红外光谱图是采用自支撑压片法得到。载体比表面积采用氮气吸附法测得,金属原子浓度则根据载体比表面积来确定。
对比例1
将拟薄水铝石(长岭催化剂厂生产的PB90粉,比表面积为345m2/g)、田菁粉按照100g:3g比例混合均匀后,加入105mL硝酸(1.5%)溶液,并再次搅拌均匀,然后将其挤条成型。经过在120℃下干燥3h和在600℃下焙烧4h步骤,制备得到粒径为1.6mm的纯氧化铝载体D1。载体D1的比表面积为283m2/g。
实施例1
将拟薄水铝石(同对比例1)、田菁粉按照100g:3g比例混合,并加入1.02g的氢氧化铍,混合均匀,加入105mL含硝酸(浓度为1.5重量%)和0.47g的HF的水溶液,并再次搅拌均匀,然后将其挤条成型。经过在120℃下干燥3h和在600℃下焙烧4h步骤,制备得到粒径为1.6mm的氧化铝载体Sup1。载体Sup1的比表面积为275m2/g。
利用红外光谱法考察上述对比例1和实施例1获得的载体的羟基振动特征,谱图如图1所示。从图1可以看出,Al2O3载体的强碱性羟基和强酸性羟基振动波数分别在3780-3750cm-1和3700-3650cm-1。相比于纯氧化铝载体D1,Sup1载体中强碱性羟基和强酸性羟基的相对高度分别为40%和35%,强碱性羟基和强酸性羟基的峰高比为1:1.14;而纯氧化铝载体D1中,强碱性羟基和强酸性羟基的峰高比为1:1.3。
对比例2
将拟薄水铝石(长岭催化剂厂生产的PB100粉,比表面积为330m2/g)、田菁粉按照100g:3.5g比例混合均匀后,加入120mL含硝酸(浓度为1.5重量%)的溶液,并再次搅拌均匀挤条成型。经过在120℃下干燥3h和在550℃下焙烧4h步骤,制备得到粒径为1.6mm的纯氧化铝载体D2。载体D2 的比表面积为292m2/g。
实施例2
将拟薄水铝石(同对比例2)、田菁粉按照100g:3.5g比例混合均匀后,加入120mL含硝酸(浓度为1.5重量%)和硼酸(1.21g)的水溶液,并再次搅拌均匀挤条成型。载体经过在120℃下干燥3h和在600℃下焙烧4h后得到氧化铝载体,焙烧后的载体以孔饱和浸渍法引入9.01g的硝酸钙,然后经过120℃干燥3h和550℃焙烧4h,得到Sup2载体。载体Sup2的比表面积为284m2/g。
利用红外光谱法考察上述载体的羟基振动特征发现,相比于纯氧化铝载体D2,Sup2中载体的强碱性羟基和强酸性羟基的相对高度分别为80%和55%,强碱性羟基和强酸性羟基的峰高比为1:0.96;而纯氧化铝载体D2中,强碱性羟基和强酸性羟基的峰高比为1:1.4。
对比例3
将拟薄水铝石(长岭催化剂厂生产的PB110粉,比表面积为325m2/g)、田菁粉按照100g:2.5g比例混合均匀后,加入120mL含硝酸(浓度为2重量%)的溶液,并再次搅拌均匀挤条成型。经过在100℃下干燥5h和在650℃下焙烧3h步骤,制备得到粒径为1.6mm的纯氧化铝载体D3。载体D3的比表面积为264m2/g。
实施例3
将拟薄水铝石(同对比例3)、田菁粉按照100g:2.5g比例混合均匀后,首先加入硅溶胶(含7.07g的SiO2)搅拌均匀,而后加入120mL的稀硝酸(浓度为2重量%),并再次搅拌均匀并挤条成型。载体经过在100℃下干燥5h 和在650℃下焙烧3h后得到氧化铝载体,焙烧后的载体以孔饱和浸渍法引入6.98g的硝酸镁,然后经过120℃干燥3h和550℃焙烧4h,得到Sup3载体。载体Sup3的比表面积为253m2/g。
利用红外光谱法考察上述对比例3和实施例3获得的载体的羟基振动特征发现,Sup3中载体的强碱性羟基和强酸性羟基的相对高度分别为70%和60%,且强碱性羟基和强酸性羟基的峰高比为1:1.07;而纯氧化铝载体D3中,强碱性羟基和强酸性羟基的峰高比为1:1.25。
实施例4
与实施例3相似,将拟薄水铝石(同实施例3)、田菁粉按照100g:2.5g比例混合均匀后,首先加入硅溶胶(含18.9g的SiO2)搅拌均匀,而后加入110mL的稀硝酸(浓度为2重量%),并再次搅拌均匀并挤条成型。载体经过在100℃下干燥5h和在650℃下焙烧3h后得到氧化铝载体,焙烧后的载体以孔饱和浸渍法引入11.64g的硝酸镁,然后经过120℃干燥3h和550℃焙烧4h,得到Sup4载体。载体Sup4的比表面积为248m2/g。
利用红外光谱法考察测得Sup4中载体的强碱性羟基和强酸性羟基的相对高度分别为30%和21%,且强碱性羟基和强酸性羟基的峰高比为1:0.88。
对比例4
按照实施例3的方法制备氧化铝载体,不同的是,硝酸镁的用量降低至1.75g量,得到对比载体D4。载体D4的比表面积为260m2/g。
利用红外光谱法考察上述对比例3和对比例4获得的载体的羟基振动特征发现,D4中载体的强碱性羟基和强酸性羟基相对高度分别为85%和95%,且强碱性羟基和强酸性羟基的峰高比为1:1.39。
对比例5
按照实施例1的方法制备氧化铝载体,不同的是,HF酸由相同重量的硝酸代替,得到对比载体D5。载体D5的比表面积为261m2/g。
利用红外光谱法考察上述对比例1和对比例5获得的载体的羟基振动特征发现,D5中载体的强碱性羟基和强酸性羟基相对高度分别为96%和52%,且强碱性羟基和强酸性羟基的峰高比为1:0.68。
对比例6
将氢氧化锆粉(比表面积为260m2/g)、田菁粉按照100g:2.5g比例混合均匀后,加入70mL的稀硝酸(浓度为2重量%),并再次搅拌均匀挤条成型。经过在100℃下干燥5h和在500℃下焙烧3h步骤,制备得到粒径为1.6mm的氧化锆载体D6。载体D6的比表面积为108m2/g。
实施例5
将氢氧化锆粉(同对比例6)、田菁粉按照100g:2.5g比例混合均匀后,首先加入含4.88g的SiO2的硅溶胶搅拌均匀,而后加入70mL的稀硝酸(浓度为2重量%),并再次搅拌均匀并挤条成型。载体经过在100℃下干燥5h和在550℃下焙烧3h后得到氧化锆载体,焙烧后的载体以孔饱和浸渍法引入4.81g的硝酸镁,然后经过120℃干燥3h和500℃焙烧4h,得到Sup5载体。载体Sup5的比表面积为102m2/g。
利用红外光谱法考察上述对比例6和实施例5获得的载体的羟基振动特征发现,Sup5中载体的强碱性羟基和强酸性羟基的相对高度分别为80%和75%,且强碱性羟基和强酸性羟基的峰高比为1:1.01;而载体D6中,强碱性羟基和强酸性羟基的峰高比为1:1.35。
实施例1-1
以Sup1为载体制备含Ni和W的催化剂Cat-Sup1,其中表面W原子浓度为3.5个原子/nm2,Ni/(Ni+W)原子比为0.3。催化剂的活性评价分别在固定床加氢脱硫评价装置上进行。原料油性质如下:密度为0.8457g/cm3,硫含量为1.1wt%,氮含量为223ppm。反应条件为:温度为325℃,压力为3.2MPa,重时空速为2h-1,氢油比为300v/v。反应在120h和240h时硫的转化率以及催化剂的脱硫转化率损失如表1所示,其中脱硫转化率损失为两个时段催化剂脱硫率差值。
对比例1-1
使用与实施例1-1相同重量的Ni、W金属活性组分并采用与实施例1-1相同的方法制备含Ni和W的催化剂Cat-D1,不同的是,载体Sup1由对比例1制得的纯氧化铝载体D1代替。然后采用与实施例1-1相同的方法进行加氢脱硫活性评价,结果如表1所示。
实施例2-1
以碱式碳酸钴、氧化钼、磷酸和乙二醇为原料,以Sup2为载体,通过饱和浸渍法制备得到Cat-Sup2催化剂。载体表面Mo原子浓度为3.2原子/nm2,Co/(Co+Mo)原子比为0.32。然后采用与实施例1-1相同的方法进行加氢脱硫活性评价,结果如表1所示。
对比例2-1
使用与实施例2-1相同重量的Co、Mo金属活性组分并采用与实施例2-1相同的方法制备含Co和Mo的催化剂Cat-D2,不同的是,载体Sup2由对比例2制得的纯氧化铝载体D2代替。然后采用与实施例1-1相同的方法进 行加氢脱硫活性评价,结果如表1所示。
实施例3-1
以碱式碳酸镍、氧化钼、偏钨酸铵、磷酸和柠檬酸为原料,以Sup3为载体,通过饱和浸渍法制备得到Cat-Sup3催化剂。载体表面W原子浓度为2.8个原子/nm2,Mo原子浓度为0.4个原子/nm2,Ni/(Ni+Mo+W)原子比为0.29。然后采用与实施例1-1相同的方法进行加氢脱硫活性评价,结果如表1所示。
对比例3-1
使用与实施例3-1相同重量的Ni、W和Mo金属活性组分并采用与实施例3-1相同的方法制备含Ni和W和Mo的催化剂Cat-D3,不同的是,载体Sup3由对比例3制得的纯氧化铝载体D3代替。然后采用与实施例1-1相同的方法进行加氢脱硫活性评价,结果如表1所示。
实施例4-1
使用与实施例3-1相同重量的Ni、W和Mo金属活性组分并采用与实施例3-1相同的方法制备含Ni和W和Mo的催化剂Cat-Sup4,不同的是,载体Sup3由实施例4制得的载体代替Sup4。然后采用与实施例1-1相同的方法进行加氢脱硫活性评价,结果如表1所示。
对比例4-1
使用与实施例3-1相同重量的Ni、W和Mo金属活性组分并采用与实施例3-1相同的方法制备含Ni和W和Mo的催化剂Cat-D4,不同的是,载体Sup3由对比例4制得的载体D4替代。然后采用与实施例1-1相同的方法进 行加氢脱硫活性评价,结果如表1所示。
对比例5-1
使用与实施例1-1相同重量的Ni、W金属活性组分并采用与实施例1-1相同的方法制备含Ni和W的催化剂Cat-D5,不同的是,载体Sup1由对比例5制得的Cat-D5载体代替。然后采用与实施例1-1相同的方法进行加氢脱硫活性评价,结果如表1所示。
对比例6-1
使用与实施例1-1相同重量的Ni、W金属活性组分并采用与实施例1-1相同的方法制备含Ni和W的催化剂Cat-D6,不同的是,载体Sup3由对比例6制得的载体D6代替。然后采用与实施例1-1相同的方法进行加氢脱硫活性评价,结果如表1所示。
实施例5-1
使用与实施例1-1相同重量的Ni、W金属活性组分并采用与实施例1-1相同的方法制备含Ni和W的催化剂Cat-Sup5,不同的是,载体Sup3由实施例5制得的载体Sup5代替。然后采用与实施例1-1相同的方法进行加氢脱硫活性评价,结果如表1所示。
实施例6-1
采用与实施例1-1相同的方法制备含Ni和W的催化剂Cat-Sup6,不同的是,调整原料的用量,使得载体表面W原子浓度为3个原子/nm2,Ni/(Ni+W)原子比为0.1。然后采用与实施例1-1相同的方法进行加氢脱硫活性评价,结果如表1所示。
表1
从表1的结果可以看出,采用本发明提供的载体并配以特定量的金属活性成分制得的催化剂的脱硫活性得到了提高,失活速度得到了抑制。由此可以看出,载体的羟基经过处理后,催化剂的脱硫活性得到了提高,失活速率得到了降低。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合。为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
- 还没有人留言评论。精彩留言会获得点赞!
- 高氯酸铵内嵌的碳/金属氧化物复合催化剂及其制备方法与制造工艺
- 一种复合氧化物负载杂多酸催化合成尿囊素的方法与制造工艺
- 一种无定形铁锌复合氧化物光芬顿催化剂的制备方法与制造工艺
- 具有杂原子的过渡金属化合物、包含该化合物的催化剂组合物和使用该催化剂组合物的聚合物的制备方法与制造工艺
- 配体化合物、过渡金属化合物和包含该化合物的催化剂组合物的制造方法与工艺
- 一种碳/金属氧化物纳米纤维复合催化剂的制备方法与制造工艺
- 改性无机耐热氧化物及其制备方法及加氢脱硫催化剂和降低加氢脱硫催化剂失活速度的方法与制造工艺
- 一种用于莫来石陶瓷晶须制备的氧化物催化剂的制造方法与工艺
- 一种中空金属蜂窝式氧化催化器的制造方法
- 一种处理汽车尾气的三元金属氧化物催化剂制备方法
- 过渡金属化合物、包含其的催化剂组合物和制备聚烯烃的方法与制造工艺
- 一种球形氮掺杂碳与过渡金属氧化物复合材料的制备方法与制造工艺
- 高氯酸铵内嵌的碳/金属氧化物复合催化剂及其制备方法与制造工艺
- 一种复合氧化物负载杂多酸催化合成尿囊素的方法与制造工艺
- 一种无定形铁锌复合氧化物光芬顿催化剂的制备方法与制造工艺
- 具有杂原子的过渡金属化合物、包含该化合物的催化剂组合物和使用该催化剂组合物的聚合物的制备方法与制造工艺
- 配体化合物、过渡金属化合物和包含该化合物的催化剂组合物的制造方法与工艺
- 烷烃氧化脱氢和/或烯烃氧化的制造方法与工艺
- 烷烃氧化脱氢和/或烯烃氧化的制造方法与工艺
- 一种碳/金属氧化物纳米纤维复合催化剂的制备方法与制造工艺
- 一种CO2加氢制甲烷整体式催化剂及其制备方法与流程
- 一种饱和加氢Pd催化剂的制备及其催化聚苯乙烯加氢的方法与流程
- 一种催化剂载体及其制备方法和用途与流程
- 用于加氢转化渣油的中孔和大孔催化剂及制备方法与制造工艺
- 一种低温等离子体改性催化剂载体的方法与制造工艺
- 一种汽油加氢脱硫催化剂及其调控制备方法与应用与制造工艺
- 碳热还原的钌碳催化剂催化衣康酸加氢制甲基丁二酸方法与制造工艺
- 改性无机耐热氧化物及其制备方法及加氢脱硫催化剂和降低加氢脱硫催化剂失活速度的方法与制造工艺
- 一种载体及其制备方法和应用以及一种催化剂及其应用以及一种加氢裂化方法与制造工艺
- 一种载体及其应用和一种催化剂及其制备方法和应用以及一种加氢裂化方法与制造工艺