唑苯衍生物及其晶体的利记博彩app
本发明涉及一种作为痛风、高尿酸血症、肿瘤溶解综合征、尿道结石、高血压、血脂异常、糖尿病、动脉硬化或心功能不全等心血管疾病、糖尿病性肾病等肾疾病、慢性阻塞性肺疾病等呼吸系统疾病、炎性肠病或自身免疫性疾病等黄嘌呤氧化酶参与的疾病的治疗药或预防药有效的新型唑苯衍生物(azolebenzenederivative)及其晶体和晶体的制备方法。
背景技术:
:黄嘌呤氧化酶是在核酸代谢中催化由次黄嘌呤向黄嘌呤转化、再向尿酸转化的酶。针对黄嘌呤氧化酶的作用,黄嘌呤氧化酶抑制剂通过抑制尿酸合成来降低血中尿酸值。即,黄嘌呤氧化酶抑制剂对高尿酸血症和由其引起的各种疾病的治疗有效。另一方面,高尿酸血症持续而导致尿酸盐晶体沉着,结果引起的病状有被称作痛风的痛风性关节炎、痛风结节。另外,高尿酸血症还作为与肥胖、高血压、血脂异常和糖尿病等有关的生活习惯病或代谢综合征的因素而受到重视,最近通过流行病学调查明确了:高尿酸血症是肾损害、尿道结石、心血管疾病的危险因素(日本痛风和核酸代谢学会指南修订委员会编,“高尿酸血症和痛风的治疗指南(高尿酸血症・痛風の治療ガイドライン)第2版”,MedicalReview社,2010)。另外,黄嘌呤氧化酶抑制剂因其对活性氧簇(reactiveoxygenspecies)产生的抑制活性而被期待着在活性氧簇参与的疾病的治疗中的有效性、例如在基于血管功能改善作用的心血管疾病治疗中的有效性(Circulation.2006;114:2508-2516)。在临床上,作为高尿酸血症的治疗药,使用别嘌醇(allopurinol)、非布索坦(febuxostat),但别嘌醇被报道有Stevens-Johnson综合征、中毒性表皮坏死症、肝损害、肾功能损害等副作用(NipponRinsho,2003;61,Suppul.1:197-201)。作为具有黄嘌呤氧化酶抑制活性的化合物,例如有人报道了2-苯基噻唑衍生物(专利文献1~3)。另一方面,在专利文献4和专利文献5中,报道了中央具有苯环的二噻唑甲酸衍生物。另外,在专利文献6和专利文献7中,报道了联苯噻唑甲酸衍生物。现有技术文献专利文献专利文献1:国际公开第92/09279号;专利文献2:日本特开2002-105067号;专利文献3:国际公开第96/31211号;专利文献4:国际公开第2011/139886号;专利文献5:国际公开第2011/101867号;专利文献6:国际公开第2010/018458号;专利文献7:国际公开第2010/128163号。技术实现要素:发明所要解决的课题本发明的目的在于提供作为痛风、高尿酸血症、肿瘤溶解综合征、尿道结石、高血压、血脂异常、糖尿病、动脉硬化或心功能不全等心血管疾病、糖尿病性肾病等肾疾病、慢性阻塞性肺疾病等呼吸系统疾病、炎性肠病或自身免疫性疾病等黄嘌呤氧化酶参与的疾病的治疗药或预防药有效的新型化合物及其晶体。另外,本发明的目的在于提供可重现性良好地制备化学上稳定、且适合作为药物原料药(原薬)的晶体的方法。用于解决课题的手段本发明人为了上述目的进行了深入地研究,结果发现了:4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸(以下,也表示为化合物(I))作为黄嘌呤氧化酶抑制剂具有优异的尿酸降低作用,可以结晶化,以及至少存在3种多晶形体。而且发现了:这些多晶形体可以通过结晶化方法分开制备。另外,本发明人发现了:4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基)-1,3-噻唑-5-甲酸钠(以下有时也表示为化合物(II))作为黄嘌呤氧化酶抑制剂具有优异的尿酸降低作用,以及化合物(II)可以结晶化。即,本发明如下:[1]4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸的晶体;[2][1]所述的晶体,其中,该晶体为A晶;[3][2]所述的晶体,其中,粉末X射线衍射光谱中,在衍射角2θ=8.6°、10.2°、13.3°、14.4°、18.5°、19.9°、21.8°、25.1°、25.6°、26.6°、27.1°和29.5°处具有特征峰;[4][2]所述的晶体,其中,粉末X射线衍射光谱具有图1所示的图案;[5][2]所述的晶体,其中,固体NMR光谱(13C)中,在化学位移为116.3ppm、117.6ppm、120.0ppm、123.6ppm、125.9ppm、127.4ppm、143.7ppm、151.8ppm、161.1ppm、162.3ppm和165.5ppm处具有特征峰;[6][2]所述的晶体,其中,固体NMR光谱(13C)具有图5所示的图案;[7][2]所述的晶体,其中,红外吸收光谱(KBr法)中,在波数为745cm-1、822cm-1、889cm-1、975cm-1、997cm-1、1611cm-1和1705cm-1处具有特征吸收峰;[8][2]所述的晶体,其中,红外吸收光谱(KBr法)具有图8所示的图案;[9][2]所述的晶体,其中,热重分析/示差热分析中的放热峰为222℃;[10][1]所述的晶体,其中,该晶体为B晶;[11][10]所述的晶体,其中,粉末X射线衍射光谱中,在衍射角2θ=10.1°、12.6°、13.1°、14.0°、18.6°、24.2°、25.2°、25.7°、27.2°和30.5°处具有特征峰;[12][10]所述的晶体,其中,粉末X射线衍射光谱具有图2所示的图案;[13][10]所述的晶体,其中,固体NMR光谱(13C)中,在化学位移为115.4ppm、118.0ppm、119.8ppm、123.2ppm、126.4ppm、129.1ppm、142.7ppm、151.2ppm、160.9ppm和166.6ppm处具有特征峰;[14][10]所述的晶体,其中,固体NMR光谱(13C)具有图6所示的图案;[15][10]所述的晶体,其中,红外吸收光谱(KBr法)中,在波数为744cm-1、810cm-1、972cm-1、997cm-1、1005cm-1、1611cm-1和1710cm-1处具有特征吸收峰;[16][10]所述的晶体,其中,红外吸收光谱(KBr法)具有图9所示的图案;[17][6]所述的晶体,其中,热重分析/示差热分析中的放热峰为225℃,为无水晶体;[18][1]所述的晶体,其中,该晶体为C晶;[19][18]所述的晶体,其中,粉末X射线衍射光谱中,在衍射角2θ=7.2°、12.5°、13.0°、14.7°、19.2°、20.0°、21.4°、21.7°、24.7°和26.0°处具有特征峰;[20][18]所述的晶体,其中,粉末X射线衍射光谱具有图3所示的图案;[21][18]所述的晶体,其中,固体NMR光谱(13C)中,在化学位移为116.1ppm、119.6ppm、123.1ppm、126.1ppm、127.1ppm、130.0ppm、143.6ppm、150.3ppm、158.3ppm、160.7ppm、163.9ppm、165.5ppm和167.0ppm处具有特征峰;[22][18]所述的晶体,其中,固体NMR光谱(13C)具有图7所示的图案;[23][18]所述的晶体,其中,红外吸收光谱(KBr法)中,在波数为745cm-1、751cm-1、809cm-1、820cm-1、971cm-1、1006cm-1、1613cm-1、1682cm-1和1710cm-1处具有特征吸收峰;[24][18]所述的晶体,其中,红外吸收光谱(KBr法)具有图10所示的图案;[25][18]所述的晶体,其中,热重分析/示差热分析中的吸热峰为88℃,放热峰为225℃;[26]4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸钠;[27][26]所述的化合物的晶体;[28][27]所述的晶体,其中,粉末X射线衍射光谱中,在衍射角2θ=7.2°、10.9°、13.3°、15.9°、18.2°、20.8°、22.1°、25.2°、26.1°和29.1°处具有特征峰;[29][27]所述的晶体,其中,粉末X射线衍射光谱具有图4所示的图案;[30][27]所述的晶体,其中,热重分析/示差热分析中的放热峰为281℃;[31]药物组合物,该药物组合物包含:[1]~[30]中任一项所述的化合物或其晶体,和药学上可接受的载体;[32]黄嘌呤氧化酶抑制药,该黄嘌呤氧化酶抑制药包含[1]~[30]中任一项所述的化合物或其晶体作为有效成分;[33]选自痛风、高尿酸血症、肿瘤溶解综合征、尿道结石、高血压、血脂异常、糖尿病、心血管疾病、肾疾病、呼吸系统疾病、炎性肠病和自身免疫性疾病的一种以上疾病的治疗药或预防药,该治疗药或预防药含有[1]~[30]中任一项所述的化合物或其晶体作为有效成分;[34]4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸的晶体(A晶)的制备方法,该制备方法包括下述的步骤:将4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸的烷基酯悬浮于溶剂中,添加碱的水溶液进行水解的步骤;和将反应物进行中和的步骤;[35][34]所述的制备方法,该制备方法进一步包括:向中和物中加入水进行搅拌的步骤;[36]4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸的晶体(B晶)的制备方法,该制备方法包括:将4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸的A晶悬浮于溶剂中的步骤;[37][36]所述的制备方法,该制备方法进一步包括:将悬浮液进行加热的步骤;[38][36]或[37]所述的制备方法,其中,溶剂为醚类、酮类、酯类、醇类、水、或它们的混合溶剂;以及[39]4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸的晶体(C晶)的制备方法,该制备方法包括:将4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸的N,N-二甲基甲酰胺溶液进行结晶。发明效果根据本发明,提供作为痛风、高尿酸血症、肿瘤溶解综合征、尿道结石、高血压、血脂异常、糖尿病、动脉硬化或心功能不全等心血管疾病、糖尿病性肾病等肾疾病、慢性阻塞性肺疾病等呼吸系统疾病、炎性肠病或自身免疫性疾病等黄嘌呤氧化酶参与的疾病的治疗药或预防药有效的唑苯衍生物的晶体及其制备方法。本发明的化合物(I)的晶体、或者化合物(II)或其晶体可以用作药物制备用原料药(原体)。另外,本发明的化合物(I)的晶体、或者化合物(II)或其晶体的制备方法为适合于工业制备的方法。附图说明图1:图1是化合物(I)的A晶的粉末X射线衍射光谱。图2:图2是化合物(I)的B晶的粉末X射线衍射光谱。图3:图3是化合物(I)的C晶的粉末X射线衍射光谱。图4:图4是化合物(II)的A晶的粉末X射线衍射光谱。图5:图5是化合物(I)的A晶的固体NMR光谱(13C)。图6:图6是化合物(I)的B晶的固体NMR光谱(13C)。图7:图7是化合物(I)的C晶的固体NMR光谱(13C)。图8:图8是化合物(I)的A晶的红外吸收光谱(KBr法)。图9:图9是化合物(I)的B晶的红外吸收光谱(KBr法)。图10:图10是化合物(I)的C晶的红外吸收光谱(KBr法)。具体实施方式“黄嘌呤氧化酶”通常以“广义”和“狭义”来使用,“广义”是指催化由次黄嘌呤向黄嘌呤转化、再向尿酸转化的氧化反应的酶,“狭义”是指作为催化同一反应的酶之一的氧化酶型的黄嘌呤氧化还原酶;在本发明中,只要没有特别说明,则“黄嘌呤氧化酶”是对催化由次黄嘌呤向黄嘌呤转化、再向尿酸转化的氧化反应的酶的总称。在承担该反应的黄嘌呤氧化还原酶中,存在氧化酶型和脱氢酶型这两种类型,这两种类型也都包含在本发明的黄嘌呤氧化酶中。在“黄嘌呤氧化酶抑制活性”、“黄嘌呤氧化酶抑制剂”等中,只要没有特别说明,则“黄嘌呤氧化酶”具有与上述定义相同的含义。本发明的晶体通过粉末X射线衍射光谱、固体NMR光谱(13C)、红外吸收光谱(KBr法)和/或热重分析/示差热分析(TG/DTA)等被表征。这些晶体的粉末X射线衍射(XRD)光谱、固体NMR光谱(13C)和红外吸收光谱(KBr法)显示特征性图案,各晶体具有特征性的衍射角2θ的值。另外,这些晶体即使在热重分析/示差热分析(TG/DTA)中也分别显示特征性的热行为。化合物(I)的A晶在粉末X射线衍射光谱中,在衍射角2θ=8.6°、10.2°、13.3°、14.4°、18.5°、19.9°、21.8°、25.1°、25.6°、26.6°、27.1°和29.5°处具有特征峰。另外,化合物(I)的A晶在粉末X射线衍射光谱中,具有图1所示的图案。化合物(I)的A晶在固体NMR光谱(13C)中,在化学位移为116.3ppm、117.6ppm、120.0ppm、123.6ppm、125.9ppm、127.4ppm、143.7ppm、151.8ppm、161.1ppm、162.3ppm和165.5ppm处具有特征峰。另外,化合物(I)的A晶在固体NMR光谱(13C)中,具有图5所示的图案。化合物(I)的A晶在红外吸收光谱(KBr法)中,在波数为745cm-1、822cm-1、889cm-1、975cm-1、997cm-1、1611cm-1和1705cm-1处具有特征吸收峰。另外,化合物(I)的A晶在红外吸收光谱(KBr法)中,具有图8所示的图案。化合物(I)的A晶在热重分析/示差热分析(TG/DTA)中,在222℃处具有放热峰。A晶为无水晶体。化合物(I)的B晶在粉末X射线衍射光谱中,在衍射角2θ=10.1°、12.6°、13.1°、14.0°、18.6°、24.2°、25.2°、25.7°、27.2°和30.5°处具有特征峰。另外,化合物(I)的B晶在粉末X射线衍射光谱中,具有图2所示的图案。化合物(I)的B晶在固体NMR光谱(13C)中,在化学位移为115.4ppm、118.0ppm、119.8ppm、123.2ppm、126.4ppm、129.1ppm、142.7ppm、151.2ppm、160.9ppm和166.6ppm处具有特征峰。另外,化合物(I)的B晶在固体NMR光谱(13C)中,具有图6所示的图案。化合物(I)的B晶在红外吸收光谱(KBr法)中,在波数为744cm-1、810cm-1、972cm-1、997cm-1、1005cm-1、1611cm-1和1710cm-1处具有特征吸收峰。另外,化合物(I)的B晶在红外吸收光谱(KBr法)中,具有图9所示的图案。化合物(I)的B晶在热重分析/示差热分析(TG/DTA)中,在225℃处具有放热峰。B晶为无水晶体。化合物(I)的C晶在粉末X射线衍射光谱中,在衍射角2θ=7.2°、12.5°、13.0°、14.7°、19.2°、20.0°、21.4°、21.7°、24.7°和26.0°处具有特征峰。另外,化合物(I)的C晶在粉末X射线衍射光谱中,具有图3所示的图案。化合物(I)的C晶在固体NMR光谱(13C)中,在化学位移为116.1ppm、119.6ppm、123.1ppm、126.1ppm、127.1ppm、130.0ppm、143.6ppm、150.3ppm、158.3ppm、160.7ppm、163.9ppm、165.5ppm和167.0ppm处具有特征峰。另外,化合物(I)的C晶在固体NMR光谱(13C)中,具有图7所示的图案。化合物(I)的C晶在红外吸收光谱(KBr法)中,在波数为745cm-1、751cm-1、809cm-1、820cm-1、971cm-1、1006cm-1、1613cm-1、1682cm-1和1710cm-1处具有特征吸收峰。另外,化合物(I)的C晶在红外吸收光谱(KBr法)中,具有图10所示的图案。化合物(I)的C晶在热重分析/示差热分析(TG/DTA)中,在88℃处具有吸热峰、在225℃处具有放热峰。C晶被认为是与二甲基甲酰胺以1:1的比例形成溶剂合物而得的晶体。化合物(II)的A晶在粉末X射线衍射光谱中,在衍射角2θ=7.2°、10.9°、13.3°、15.9°、18.2°、20.8°、22.1°、25.2°、26.1°和29.1°处具有特征峰。另外,化合物(II)的A晶在粉末X射线衍射光谱中,具有图4所示的图案。在热重分析/示差热分析(TG/DTA)中,在281℃处具有放热峰。A晶为无水晶体。在此,“特征峰”是指,在各多晶形的粉末X射线衍射光谱、固体NMR光谱(13C)和红外吸收光谱(KBr法)中主要观察到的峰和固有的峰。通过本发明的衍射角特别规定的晶体中也包含观察到上述特征峰以外的峰的晶体。粉末X射线衍射光谱中的衍射角2θ的位置和相对强度可根据测定条件而略微变动,因此,即使在2θ略微不同的情况下,也应该适当参照光谱整体的图案来认定晶形的同一性,这样的误差范围的晶体也包括在本发明中。作为2θ的误差,例如认为是±0.5°、±0.2°。即,通过上述衍射角特别规定的晶体还包含在±0.5°或±0.2°的范围内一致的晶体。一般来说,固体NMR光谱(13C)中的化学位移也可产生误差。作为这样的误差,例如为±0.25ppm、代表性的为±0.5ppm的范围。即,通过上述化学位移特别规定的晶形还包含在±0.25ppm或±0.5ppm的范围内一致的晶体。一般来说,红外吸收光谱(KBr法)中的吸收峰也可产生误差。作为这样的误差,例如为±2cm-1、代表性的为±5cm-1的范围。即,通过上述波数特别规定的晶形还包括在±2cm-1或±5cm-1的范围内一致的晶体。在热重分析/示差热分析(TG/DTA)中,“放热峰”和“吸热峰”是峰的起始点的温度,是指通过外推求出的放热和吸热的起始温度。TG/DTA中的吸热和放热峰也可根据测定条件而略微变动。作为误差,例如认为是±5℃、±2℃的范围。即,通过上述峰特别规定的晶体还包含±5℃或±2℃的范围内一致的晶体。并且,关于粉末X射线衍射光谱、固体13CNMR光谱、红外吸收光谱(KBr法)、TG/DTA的任一种,对于晶体的标准品、例如通过本申请实施例记载的方法获得的各晶体的实测值与本申请记载的数值之差也允许作为测定误差。即,在通过这样的方法算出的误差范围内,衍射角、化学位移、赤外吸收峰、或吸热和放热峰一致的晶体也包含在本发明的晶体中。化合物(I)的A晶可通过下述的方法制备,所述方法包含下述的步骤:将化合物(I)的烷基酯悬浮于溶剂中,添加碱的水溶液进行水解的步骤;以及,将反应物进行中和的步骤。另外,化合物(I)的A晶的制备方法可以进一步包含下述的步骤:向中和物中添加水的步骤;接着,将反应溶液进行搅拌的步骤。作为将化合物(I)的烷基酯悬浮的溶剂,例如可以举出:苯、甲苯、二甲苯等芳族烃类;二乙醚、四氢呋喃(THF)、1,4-二噁烷、1,2-二甲氧基乙烷、1,2-二乙氧基乙烷、甲基叔丁基醚等醚类;二氯甲烷、1,2-二氯乙烷、氯仿等卤代烃类;甲醇、乙醇、2-丙醇、丁醇、六氟-2-丙醇、三氟乙醇等醇类;N,N-二甲基甲酰胺(DMF)、N,N-二甲基乙酰胺(DMA)、N-甲基吡咯烷酮、二甲基亚砜(DMSO)、乙腈、丙酮、乙酸乙酯、水或它们的混合溶剂等。优选为醚类或醇类、水或它们的混合溶剂。作为化合物(I)的烷基酯,优选碳原子数1~6的烷基酯,更优选乙酯。在此,烷基酯是指直链或支链的脂肪族饱和烃酯。作为碳原子数1~6的烷基酯,可以举出:甲酯、乙酯、异丙酯、叔丁酯等作为具体例子。由化合物(I)的烷基酯水解成化合物(I)的反应,可通过将化合物(I)的烷基酯悬浮于上述溶剂(例如,烷基酯的15倍量)中之后,使等量或稍微过量的碱与化合物(I)的烷基酯反应来进行。作为优选的碱,可以举出:氢氧化钠、氢氧化钾、氢氧化锂。本反应在0℃~100℃下进行,优选为20~30℃。水解反应后,通过使与所用的碱等量或稍微过量的酸发生反应来进行中和。作为优选的酸,可以举出盐酸。中和反应可在0℃~100℃下进行,优选为0℃~30℃。接着,向中和了的反应物中加入水(例如,烷基酯的5倍量),搅拌1小时之后,滤取析出物并干燥,获得晶体。对溶剂量、水的添加量、搅拌条件、至过滤分离(濾別)为止的时间没有特别限定,但这些条件有时会影响晶体的收率、化学纯度、粒径、粒度分布等,因此,优选根据目的来组合设定。滤取可以使用通常的方法,例如自然过滤、加压过滤、减压过滤、或离心分离。干燥可以使用通常的方法,例如自然干燥、减压干燥、加热干燥、减压加热干燥。化合物(I)的烷基酯的合成可通过任何方法来进行,例如,可以通过以下的方法来合成。化合物(A-2)的合成[化学式1](式中,Y1和Y2表示离去基团。)作为Y1和Y2所示的离去基团,可以举出:卤原子、甲磺酰氧基、对甲苯磺酰氧基、三氟甲磺酰氧基等。本反应是通过在碱存在下使具有离去基团的烷基化试剂与化合物(A-1)中的酚式羟基反应来合成化合物(A-2)的方法。作为所使用的碱,可以使用:氢化钠、氢氧化钠、氢氧化钾、氢氧化锂、碳酸钠、碳酸钾、碳酸铯等无机盐;乙醇钠、甲醇钠、叔丁氧基钾等金属醇盐;三乙胺、吡啶、4-氨基吡啶、N-乙基-N,N-二异丙胺(DIPEA)、1,8-二氮杂双环[5.4.0]-7-十一碳烯(DBU)等有机胺。本反应通过如下进行:在0℃~140℃下,在对反应呈惰性的溶剂中,使等量或稍微过量的碱与化合物(A-1)反应之后,加入等量或过量的烷基化试剂反应通常0.5~16小时。本反应优选在氮等惰性气体气氛下进行。在此,作为溶剂没有特别限定,例如可以举出:二乙醚、四氢呋喃(THF)、1,4-二噁烷、1,2-二甲氧基乙烷、1,2-二乙氧基乙烷等醚类;N,N-二甲基甲酰胺(DMF)、N-甲基吡咯烷酮、二甲基亚砜(DMSO)、水或它们的混合溶剂等。化合物(A-4)的合成[化学式2](式中,R表示碳原子数1~6的烷基。)本合成法是通过使化合物(A-2)和(A-3)偶联来合成化合物(A-4)的方法。作为Y1所示的离去基团,可以举出:卤原子、甲磺酰氧基、对甲苯磺酰氧基、三氟甲磺酰氧基等。本反应通过如下进行:使用等量或一方过量的化合物(A-2)和(A-3),在对反应呈惰性的溶剂中,在碱和过渡金属催化剂存在下,视情况加入配体、羧酸和铜(I价或II价)盐,在室温~加热回流下通常反应0.5小时~2天。本反应优选在氮等惰性气体气氛下进行。在此,作为溶剂没有特别限定,例如可以举出:苯、甲苯、二甲苯等の芳族烃类、;二乙醚、四氢呋喃(THF)、1,4-二噁烷、1,2-二甲氧基乙烷、1,2-二乙氧基乙烷等醚类;二氯甲烷、1,2-二氯乙烷、氯仿等卤代烃类;甲醇、乙醇、2-丙醇、丁醇等醇类;N,N-二甲基甲酰胺(DMF)、N-甲基吡咯烷酮、二甲基亚砜(DMSO)、水或它们的混合溶剂等。作为碱,可以举出:氢化锂、氢化钠、氢化钾、氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸铯、氟化钾、氟化铯、磷酸三钾、乙酸钠、乙酸钾等,碳原子数1~6的醇盐的金属盐(锂盐、钠盐、钾盐、镁盐)、碳原子数1~6的烷基阴离子的金属盐(锂盐、钠盐、钾盐、镁盐)、四(碳原子数1~4的烷基)铵盐(氟化盐、氯化盐、溴化盐)、二异丙基乙胺、三丁胺、N-甲基吗啉、二氮杂双环十一碳烯、二氮杂双环辛烷或咪唑等。作为过渡金属催化剂,可以举出:铜、钯、钴、铁、铑、钌和铱等。作为配体,可以举出:三(叔丁基)膦、三(环己基)膦、叔丁基二环己基膦、二(叔丁基)环己基膦或二(叔丁基)甲基膦等。作为铜(I价或II价)盐,可以举出:氯化铜(I)、溴化铜(I)、碘化铜(I)、乙酸铜(I)、氟化铜(II)、氯化铜(II)、溴化铜(II)、碘化铜(II)、乙酸铜(II)和它们的水合物、以及它们的混合物等。作为羧酸,可以举出:甲酸、乙酸、丙酸、正丁酸、异丁酸、戊酸、异戊酸、特戊酸和三氟乙酸等。化合物(A-5)的合成[化学式3](式中,R表示碳原子数1~6的烷基。)本合成法是通过将化合物(A-4)的硝基还原来合成化合物(A-5)的方法。本反应通过如下进行:将化合物(A-4)在对反应呈惰性的溶剂中,在过渡金属催化剂存在下,在氢气气氛下于室温~加热回流下通常反应0.5小时~2天。在此,作为溶剂没有特别限定,例如可以举出:苯、甲苯、二甲苯等芳族烃类;二乙醚、四氢呋喃(THF)、1,4-二噁烷、1,2-二甲氧基乙烷、1,2-二乙氧基乙烷等醚类;二氯甲烷、1,2-二氯乙烷、氯仿等卤代烃类;甲醇、乙醇、2-丙醇、丁醇等醇类;N,N-二甲基甲酰胺(DMF)、N-甲基吡咯烷酮、二甲基亚砜(DMSO)、乙酸乙酯或它们的混合溶剂等。作为过渡金属催化剂,优选钯-碳、氢氧化钯、钯黑、铂-碳、拉尼镍等。化合物(A-6)的合成[化学式4](式中,R和R1独立表示碳原子数1~6的烷基。)本合成法是使化合物(A-5)和原甲酸酯和叠氮化合物反应来合成四唑环的方法。本反应通过如下进行:使用等量或任一方过量的化合物(A-5)、原甲酸酯和叠氮化合物,在对反应呈惰性的溶剂中,在酸存在下于室温~加热回流下通常反应0.5小时~2天。本反应优选在氮等惰性气体气氛下进行。作为原甲酸酯,可以举出:原甲酸三甲酯和原甲酸三乙酯等。另外,作为叠氮化合物,可以举出:叠氮化钠、三甲基甲硅烷基叠氮化物等。作为所使用的酸,可以举出:甲酸、乙酸等有机酸;盐酸、硫酸等无机酸;三氟甲磺酸铟、三氟甲磺酸镱、三氟甲磺酸锌、三氯化铟等路易斯酸。作为用于这些反应的溶剂,没有特别限定,例如可以举出:苯、甲苯、二氯甲烷、二氯乙烷、氯仿、四氯化碳、二乙醚、四氢呋喃(THF)、1,4-二噁烷、1,2-二甲氧基乙烷、1,2-二乙氧基乙烷、N,N-二甲基甲酰胺(DMF)、N-甲基吡咯烷酮、二甲基亚砜(DMSO)或它们的混合溶剂等,也可以将乙酸等的酸用作溶剂。在合成过程中,如果需要,可以通过重结晶、再沉淀或各种色谱法等常规方法纯化上述的中间体化合物。化合物(I)的B晶可以通过包括将化合物(I)的A晶悬浮于溶剂的步骤的方法进行制备。另外,化合物(I)的B晶的制备方法可以进一步包括:接着将反应溶液进行加热的步骤。在化合物(I)的B晶的制备中,作为将A晶悬浮的溶剂,例如可以举出:苯、甲苯、二甲苯等芳族烃类;二乙醚、四氢呋喃(THF)、1,4-二噁烷、1,2-二甲氧基乙烷、1,2-二乙氧基乙烷、甲基叔丁基醚等醚类;丙酮、2-丁酮等酮类;乙酸乙酯、乙酸异丁酯等酯类;二氯甲烷、1,2-二氯乙烷、氯仿等卤代烃类;甲醇、乙醇、2-丙醇、丁醇、六氟-2-丙醇、三氟乙醇等醇类;N,N-二甲基甲酰胺(DMF)、N,N-二甲基乙酰胺(DMA)、N-甲基吡咯烷酮、二甲基亚砜(DMSO)、乙腈、丙酮、乙酸乙酯、甲基乙基酮、水或它们的混合溶剂等。优选为醚类、酮类、酯类、醇类、水或它们的混合溶剂。将化合物(I)的A晶转换为化合物(I)的B晶通过如下进行:使化合物(I)的A晶悬浮于上述溶剂(例如,化合物(I)的A晶的5~60倍量)中之后,使反应溶液加热回流6小时。接着,将反应溶液在25℃下搅拌之后,滤取析出物并干燥,获得晶体。溶剂量、加热时间、搅拌条件、至过滤分离为止的时间没有特别限定,但这些条件有时会影响晶体的收率、化学纯度、粒径、粒度分布等,因此,优选根据目的来组合设定。滤取可以使用通常的方法,例如自然过滤、加压过滤、减压过滤、或离心分离。干燥可以使用通常的方法,例如自然干燥、减压干燥、加热干燥、减压加热干燥。化合物(I)的C晶可以通过将化合物(I)利用N,N-二甲基甲酰胺溶剂进行结晶来获得。向化合物(I)中加入N,N-二甲基甲酰胺(例如,化合物(I)的10倍量),在80℃下加热搅拌使之溶解。使反应溶液冷却至20~30℃,搅拌2小时。滤取析出物,将过滤物用乙醇(例如,化合物(I)的10倍量)洗涤。将母液在20~30℃下静置7天,滤取析出物并干燥,获得晶体。对溶剂、搅拌条件、至过滤分离为止的时间没有特别限定,但这些条件有时影响晶体的收率、化学纯度、粒径、粒度分布等,因此,优选根据目的来组合设定。滤取可以使用通常的方法,例如自然过滤、加压过滤、减压过滤、或离心分离。干燥可以使用通常的方法,例如自然干燥、减压干燥、加热干燥、减压加热干燥。另外,化合物(I)的C晶还可以通过包括将化合物(I)的B晶悬浮于溶剂中的步骤的方法进行制备。向化合物(I)的B晶中加入N,N-二甲基甲酰胺和乙酸乙酯的1:1的混合溶液(例如,化合物(I)的10倍量),在室温下使其搅拌14天。过滤溶液,将过滤物在室温下干燥,获得晶体。溶剂、搅拌条件、至过滤分离为止的时间没有特别限定,但这些条件有时影响晶体的收率、化学纯度、粒径、粒度分布等,因此,优选根据目的来组合设定。滤取可以使用通常的方法,例如自然过滤、加压过滤、减压过滤、或离心分离。干燥可以使用通常的方法,例如自然干燥、减压干燥、加热干燥、减压加热干燥。化合物(I)可以通过例如以下的方法来合成。[化学式5](式中,R2表示羧基的保护基。)本合成法是通过将化合物(A-7)的保护基R2利用酸或碱等脱保护来合成化合物(I)的方法。“羧基的保护基”是指,例如PROTECTIVEGROUPSinORGANICSYNTHESIS(有机合成中的保护基),第三版,JohnWiley&Sons,Inc.中记载的常规的羧基保护基,例如可以举出:甲基、乙基、异丙基、庚基、叔丁基、甲氧基甲基、甲硫基甲基、甲氧基乙氧基甲基、甲氧基乙基、苄基等。化合物(A-7)可以通过将上述化合物(A-6)的合成方法中的R置换为R2的方法来合成。本反应通过如下进行:对化合物(A-7),在对反应呈惰性的溶剂中使用等量或过量的酸或碱,于室温~加热回流下通常反应0.5小时~5天。在此,作为溶剂没有特别限定,例如可以举出:苯、甲苯、二甲苯等芳族烃类;二乙醚、四氢呋喃(THF)、1,4-二噁烷、1,2-二甲氧基乙烷、1,2-二乙氧基乙烷等醚类;二氯甲烷、1,2-二氯乙烷、氯仿等卤代烃类;甲醇、乙醇、2-丙醇、丁醇等醇类;N,N-二甲基甲酰胺(DMF)、N-甲基吡咯烷酮、二甲基亚砜(DMSO)、水或它们的混合溶剂等。作为酸,可以举出:氯化氢、溴化氢、硫酸、硝酸、磷酸等无机酸;或将这些酸用水或有机溶剂稀释而得的溶液等。作为碱,可以举出:氢氧化钠、氢氧化钾、氢氧化锂、碳酸钠、碳酸钾等无机碱;乙醇钠、甲醇钠等金属醇盐;或将这些碱用水或有机溶剂稀释而得的溶液等。脱保护中使用碱时,通过在反应后加入酸进行中和,获得化合物(I)。作为用于中和的酸,可以使用前面例示的酸。如果需要,可以通过重结晶、再沉淀或各种色谱法等常规方法纯化所得的化合物(I)。化合物(II)的A晶可以通过例如以下的方法来合成。[化学式6]化合物(II)可以通过包括将化合物(I)悬浮于溶剂中并添加氢氧化钠的步骤的方法进行制备。另外,可以进一步包括将反应溶液进行搅拌的步骤。作为将化合物(I)悬浮的溶剂,例如可以举出:苯、甲苯、二甲苯等芳族烃类;二乙醚、四氢呋喃(THF)、1,4-二噁烷、1,2-二甲氧基乙烷、1,2-二乙氧基乙烷等醚类;二氯甲烷、1,2-二氯乙烷、氯仿等卤代烃类;甲醇、乙醇、2-丙醇、丁醇等醇类;N,N-二甲基甲酰胺(DMF)、N-甲基吡咯烷酮、二甲基亚砜(DMSO)、水或它们的混合溶剂等。优选为醚类或醇类、水或它们的混合溶剂。由化合物(I)到化合物(II)的盐形成反应通过如下进行:将化合物(I)悬浮于上述溶剂(例如,羧酸的10倍量)中之后,使等量或稍微过量的氢氧化钠与化合物(I)反应。本反应在0℃~100℃下进行,优选为0~30℃。接着,搅拌1小时之后,滤取析出物并干燥,获得晶体。溶剂量、搅拌条件、至过滤分离为止的时间没有特别限定,但这些条件有时影响晶体的收率、化学纯度、粒径、粒度分布等,因此,优选根据目的来组合设定。滤取可以使用通常的方法,例如自然过滤、加压过滤、减压过滤、或离心分离。干燥可以使用通常的方法,例如自然干燥、减压干燥、加热干燥、减压加热干燥。在合成过程中,如果需要,可以通过重结晶、再沉淀或各种色谱法等常规方法纯化上述合成法中的中间体化合物。本发明的各晶体可以通过特征性粉末X射线衍射光谱、固体NMR光谱(13C)、红外吸收光谱(KBr法)或热重分析/示差热分析(TG/DTA)来与其它晶形加以区分,但不涉及其它晶形的混入率这一方面。单独获得特定的晶体时,至少,如果是通过这些测定方法无法检出的程度的混入,则可允许。另外,作为药物而将各特定的晶体用作原料药时,并不表示不允许含有其它晶体。本发明的各晶体均可用作药物的有效成分。另外,不仅可以使用单独的晶体,也可以使用2种以上的混合物。本发明中,化合物(I)和化合物(II)的晶体与非晶相比,制备时的操作、重现性、稳定性以及保存稳定性等变得有利。使用本发明的化合物(I)的晶体、或者化合物(II)或其晶体和药物上可接受的载体,能够制成药物组合物。含有本发明的化合物(I)的晶体、或者化合物(II)或其晶体的制剂可以使用通常用于制剂化的添加剂来调制。作为这些添加剂,在固体制剂的情况下,可以举出:乳糖、白糖、葡萄糖、玉米淀粉、马铃薯淀粉、结晶纤维素、轻质硅酸酐、合成硅酸铝、偏硅酸铝酸镁和磷酸氢钙等赋形剂;结晶纤维素、羧甲基纤维素、羟丙基纤维素、羧甲基纤维素钠和聚乙烯基吡咯烷酮等粘合剂;淀粉、羧甲基纤维素钠、羧甲基纤维素钙、交联羧甲基纤维素钠和羧甲基淀粉钠等崩解剂;滑石和硬脂酸类等润滑剂;羟甲丙纤维素、羟丙甲纤维素邻苯二甲酸酯和乙基纤维素等涂覆剂;着色剂。在半固体制剂的情况下,可以举出:白色凡士林等主剂。在液体制剂的情况下,可以举出:乙醇等溶剂;乙醇等溶解助剂;对羟基苯甲酸酯类等保存剂;葡萄糖等等张化剂;柠檬酸类等缓冲剂;L-抗坏血酸等抗氧化剂;EDTA等螯合剂;和聚山梨醇酯80等悬浮剂/乳化剂等。本发明的化合物(I)的晶体、或者化合物(II)或其晶体即使是固体制剂、半固体制剂和液体制剂等的任意剂型,口服制剂和肠胃外制剂(注射剂、经皮剂、滴眼剂、栓剂、经鼻剂和吸入剂等)中的任意应用制剂均可以使用。含有本发明的化合物(I)的晶体、或者化合物(II)或其晶体作为有效成分的药物组合物可以用作黄嘌呤氧化酶抑制药,或痛风、高尿酸血症、肿瘤溶解综合征、尿道结石、高血压、血脂异常、糖尿病、动脉硬化或心功能不全等心血管疾病、糖尿病性肾病等肾疾病、慢性阻塞性肺疾病等呼吸系统疾病、炎性肠病或自身免疫性疾病等黄嘌呤氧化酶参与的疾病的治疗药或预防药。在此,“预防”是指针对尚未患病或发病的个体将患病或发病防范于未然,“治疗”是指针对已经患病或发病的个体治愈、抑制或改善疾病或症状。实施例[测定方法]本发明的晶体的粉末X射线衍射利用以下条件进行了测定。装置:BrukerAXS制造的D8DISCOVERWithGADDSCS射线源:CuKα;波长:1.541838(10-10m)、40kv-40mA、入射侧平板石墨单色器、准直管φ300μm、二维PSPC检测器、扫描3~40°。[测定方法]本发明的晶体的固体NMR光谱(13C)利用以下条件进行了测定。装置:Bruker制造的DSX300WB测定核:13C脉冲重复时间:5秒脉冲模式:CP/MAS测定。[测定方法]本发明的晶体的红外吸收光谱(KBr法)按照日本药典的一般试验法中记载的红外吸收光谱测定法的溴化钾片剂法,利用以下条件进行了测定。装置:ThermoFischerScientific制造的AVATAR320测定范围:4000~400cm-1分辨率:4cm-1累计次数:64。本发明的晶体的热重分析/示差热分析(TG/DTA)利用以下条件进行了测定。装置:Rigaku制造的TG8120升温速度:每分钟10℃,气氛:氮气,样品盘:铝,对照:氧化铝,取样:1.0秒,测定温度范围:25~300℃对于测定了1HNMR光谱(400MHz、DMSO-d6或CDCl3)的化合物,示出其化学位移(δ:ppm)和耦合常数(J:Hz)。下述简写分别表示以下的含义。装置:JEOL制造的JMTC-400/54/SSs=单峰、d=双峰、t=三重峰、q=四重峰、brs=宽单峰、m=多重峰。[参考例1]4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸乙酯的制备(1)使20.0g的4-溴-2-硝基苯酚和19.0g碳酸钾悬浮于200mL二甲基甲酰胺中,在氮气氛下加入20.1g异丁基溴,在110℃下加热了22小时。向反应混合液中加入水,用乙酸乙酯进行了萃取。将有机层用食盐水洗涤后,进行干燥、减压浓缩,获得了24.8g的4-溴-1-(2-甲基丙氧基)-2-硝基苯。(2)向2.74g的4-溴-1-(2-甲基丙氧基)-2-硝基苯中加入2.10g碳酸氢钾、43.5mg氯化钯(II)、205mg溴化铜(I),使其悬浮于35mL甲苯中。然后,加入2.05g的4-甲基-1-1,3-噻唑-5-甲酸乙酯、92μL异丁酸和230μL二-叔丁基环己基膦,在氮气氛下、120℃下加热了14小时。将反应混合液进行硅藻土(celite)过滤,去除不溶物,向滤液中加入水,用乙酸乙酯进行了萃取。将有机层用食盐水洗涤后,进行干燥、减压浓缩之后,利用常规方法进行纯化,获得了3.16g的4-甲基-2-[3-硝基-4-(2-甲基丙氧基)苯基]-1,3-噻唑-5-甲酸乙酯。(3)使3.16g的4-甲基-2-[3-硝基-4-(2-甲基丙氧基)苯基]-1,3-噻唑-5-甲酸乙酯悬浮于30mL乙醇中,加入200mg钯/碳(10%wt)之后,在氢气氛下、50℃下搅拌了14小时。过滤反应混合液,将滤液进行减压浓缩,由此获得了2.12g的2-[3-氨基-4-(2-甲基丙氧基)苯基]-4-甲基-1,3-噻唑-5-甲酸乙酯。(4)使2.12g的2-[3-氨基-4-(2-甲基丙氧基)苯基]-4-甲基-1,3-噻唑-5-甲酸乙酯悬浮于20mL乙酸中之后,加入780mg叠氮化钠、1.78g原甲酸三乙酯,在氮气氛下、70℃下加热了2小时。冷却至室温之后向反应混合液中加入20mL水,利用常规方法进行纯化,获得了2.25g的4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸乙酯。[实施例1]4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸(化合物(I))的A晶的制备将883g的4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸乙酯溶解于13.2L四氢呋喃/甲醇=1/1的混合溶液中,加入2.28L的2M氢氧化钠水溶液,在20~30℃下搅拌了3小时。在20~30℃下边搅拌边向反应混合液中缓慢加入2.28L的2M盐酸,进一步缓慢加入了4.4L水。将反应溶液在20~30℃下搅拌1小时之后,滤取了晶体。将所得的晶体用4.4L甲醇/水=1/1的混合溶剂和4.4L水进行了洗涤。将晶体在50℃下减压干燥,获得了811g的4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸的晶体。所得晶体的XRD示于图1。在衍射角2θ=8.6°、10.2°、13.3°、14.4°、18.5°、19.9°、21.8°、25.1°、25.6°、26.6°、27.1°和29.5°处观测到了峰。所得晶体的固体NMR光谱(13C)示于图5。在化学位移为116.3ppm、117.6ppm、120.0ppm、123.6ppm、125.9ppm、127.4ppm、143.7ppm、151.8ppm、161.1ppm、162.3ppm和165.5ppm处观测到了峰。所得晶体的红外吸收光谱(KBr法)示于图8。在波数为745cm-1、822cm-1、889cm-1、975cm-1、997cm-1、1611cm-1和1705cm-1处观测到了峰。另外,热重分析/示差热分析(TG/DTA)中的放热峰为222℃。[实施例2]4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸(化合物(I))的B晶的制备使3.0g化合物(I)的A晶悬浮于21.0mL乙酸乙酯中,回流了6小时。冷却至25℃之后在25℃下搅拌了30分钟。滤取晶体,用15mL乙酸乙酯进行了洗涤。将晶体在45℃下减压干燥,获得了2.9g的4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸的晶体。所得晶体的XRD示于图2。在衍射角2θ=10.1°、12.6°、13.1°、14.0°、18.6°、24.2°、25.2°、25.7°、27.2°和30.5°处观测到了峰。所得晶体的固体NMR光谱(13C)示于图6。在化学位移为115.4ppm、118.0ppm、119.8ppm、123.2ppm、126.4ppm、129.1ppm、142.7ppm、151.2ppm、160.9ppm和166.6ppm处观测到了峰。所得晶体的红外吸收光谱(KBr法)示于图9。在波数为744cm-1、810cm-1、972cm-1、997cm-1、1005cm-1、1611cm-1和1710cm-1处观测到了峰。另外,热重分析/示差热分析(TG/DTA)中的放热峰为225℃。[参考例2]4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸(化合物(I))的制备将4.00g的4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸乙酯溶解于60mL四氢呋喃/甲醇=1/1的混合溶液中,加入10.3mL的2M氢氧化钠水溶液,在室温下搅拌了3小时。向反应混合液中加入10.3mL的2M盐酸进行搅拌之后,加入60mL水,利用常规方法进行纯化,获得了3.50g的4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸。[实施例3]4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸(化合物(I))的C晶的制备向8.5g的4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸中加入90mL的N,N-二甲基甲酰胺,通过在80℃下加热搅拌进行了溶解。冷却至室温后,在室温下搅拌2小时之后,进行过滤,将过滤物用90mL乙醇进行了洗涤。将母液在室温下静置7天,所得的晶体用30mL乙醇进行了洗涤。将晶体在40℃下减压干燥,获得了1.0g化合物(I)的晶体。所得晶体的XRD示于图3。在衍射角2θ=7.2°、12.5°、13.0°、14.7°、19.2°、20.0°、21.4°、21.7°、24.7°和26.0°处观测到了峰。所得晶体的固体NMR光谱(13C)示于图7。在化学位移为116.1ppm、119.6ppm、123.1ppm、126.1ppm、127.1ppm、130.0ppm、143.6ppm、150.3ppm、158.3ppm、160.7ppm、163.9ppm、165.5ppm和167.0ppm处观测到了峰。所得晶体的红外吸收光谱(KBr法)示于图10。在波数为745cm-1、751cm-1、809cm-1、820cm-1、971cm-1、1006cm-1、1613cm-1、1682cm-1和1710cm-1处观测到了峰。另外,热重分析/示差热分析(TG/DTA)中的吸热峰为88℃、放热峰为225℃。[实施例4]4-甲基-2-[4-(2-甲基丙氧基)-3-(1H-1,2,3,4-四唑-1-基)苯基]-1,3-噻唑-5-甲酸钠(化合物(II))的A晶的制备将400mg氢氧化钠溶解于36mL乙醇中,加入3.59g化合物(I),在20~30℃下搅拌了1小时。然后,滤取了晶体。所得的晶体用10mL乙醇进行了洗涤。将晶体在50℃下减压干燥,获得了3.53g化合物(II)的晶体。所得晶体的XRD示于图4。在衍射角2θ=7.2°、10.9°、13.3°、15.9°、18.2°、20.8°、22.1°、25.2°、26.1°和29.1°处观测到了峰。另外,热重分析/示差热分析(TG/DTA)中的放热峰为281℃。[实施例5]血中尿酸降低作用(正常大鼠)确认了化合物(II)的血中尿酸降低作用。使用进食针(feedingneedle)对8~9周龄的Sprague-Dawley系雄性大鼠(日本CharlesRiver株式会社)强制给予了悬浮于0.5%甲基纤维素溶液中的试验化合物。给予后2小时,从尾静脉采血,之后分离了血浆。使用尿酸测定试剂盒(L型WakoUAF:和光纯药工业),按照尿酸酶法,利用吸光度计测定血中尿酸值,通过下式求出了尿酸降低率。尿酸降低率(%)=(对照动物的尿酸值-给予试验化合物的动物的尿酸值)×100/对照动物的尿酸值化合物(II)的A晶在10mg/kg、1mg/kg的任一种的用量下,均显示出尿酸降低率为50%以上。由该结果显示出:化合物(II)具有强力的血中尿酸降低效果。[实施例6]血中尿酸降低作用的持续性(正常大鼠)将化合物(II)的A晶利用与实施例5同样的方法给予了Sprague-Dawley系雄性大鼠。在给予后24小时,从尾静脉采血,之后分离了血浆。使用尿酸测定试剂盒(L型WAKOUAF:和光纯药工业),按照尿酸酶法,利用吸光度计测定血中尿酸值,通过下式求出了尿酸降低率。尿酸降低率(%)=(对照动物的尿酸值-给予试验化合物的动物的尿酸值)×100/对照动物的尿酸值化合物(II)的A晶在给予后24小时,在10mg/kg的用量下,显示出尿酸降低率为50%以上,在3mg/kg的用量下,显示出尿酸降低率为40%以上。由该结果显示出:化合物(II)具有长期、持续的血中尿酸降低效果。[实施例7]在JP2溶液中的溶解性确认了化合物(II)在JP2溶液(pH6.8)中的溶解性。将化合物(II)的A晶在乳钵中轻轻地粉碎,向20mL的JP2溶液中添加约4mg,在室温下进行了搅拌。对于该溶液,从刚添加后起,通过紫外可见吸收度测定法相继地测定280nm下的吸光度(At),以预先测定的标准溶液的280nm下的吸光度(As)为指标,通过下式求出了各测定时间下的溶解度。测定持续至120分钟为止。溶解度=At/As×Cs(其中,Cs表示标准溶液的浓度)测定条件测定装置:Pion公司制造μDISSProfiler搅拌速度:700rpmUV测定波长:280nm测定间隔:测定开始~1分钟:3秒1~8分钟:20秒8~120分钟:2分钟在添加后30秒以内,化合物(II)的A晶所添加的总量溶解,溶解度为187μg/mL。另外,添加后120分钟的溶解度为189μg/mL,未观察到析出,维持了溶解状态。由该结果显示出:化合物(II)具有优异的溶解性。[参考例3]黄嘌呤氧化酶抑制活性的测定(1)试验化合物的调制将化合物(I)溶解于DMSO(Sigma公司制造)中达到20mM的浓度,之后,使用时调制成目标浓度进行使用。(2)测定方法化合物(I)的黄嘌呤氧化酶抑制活性评价通过将文献记载的方法(MethodEnzymaticAnalysis(方法酶法分析),1,521-522,1974)部分改变来进行实施。本评价通过测定氧化酶型的黄嘌呤氧化还原酶活性来进行。即,将预先通过20mM氢氧化钠溶液调制成10mM的黄嘌呤(Sigma公司制造)溶液使用100mM磷酸缓冲液调制成30μM,以每孔75μL加入到96孔板中。将通过DMSO稀释达到最终浓度的100倍的试验化合物以每孔1.5μL进行添加,混合后利用微孔板读板仪SPECTRAMAXPlus384(MolecularDevices公司制造)测定了290nm的吸光度。接着,将氧化酶型黄嘌呤氧化还原酶(来自酪乳、Calbiochem公司制造)使用100mM磷酸缓冲液调制成30.6mU/mL,以每孔73.5μL进行了添加。混合后迅速测定了5分钟的290nm下的吸光度变化。将代替试验化合物溶液添加了DMSO时的酶活性作为100%,计算试验化合物的抑制率,使其与剂量应答曲线拟合,计算了对氧化酶型黄嘌呤氧化还原酶的50%抑制浓度。化合物(I)显示出1.0nM≦IC50<5.0nM的黄嘌呤氧化酶抑制活性。[参考例4]血中尿酸降低作用(正常大鼠)对于化合物(I),利用与实施例5同样的方法,确认了血中尿酸降低作用。化合物(I)在10mg/kg、1mg/kg的任一种的用量下,均显示出尿酸降低率为50%以上。由该结果显示出:化合物(I)具有强力的血中尿酸降低效果。[参考例5]血中尿酸降低作用的持续性(正常大鼠)对于化合物(I),利用与实施例6同样的方法,确认了血中尿酸降低作用的持续性。化合物(I)在给予后24小时,在10mg/kg的用量下,显示出尿酸降低率为50%以上,在3mg/kg的用量下,显示出尿酸降低率为40%以上。由该结果显示出:化合物(I)具有长期、持续的血中尿酸降低效果。[参考例6]血中尿酸降低作用(高尿酸血症比格(beagle)犬)对于化合物(I),确认了由氧嗪酸(oxonicacid)诱发了高尿酸血症的比格犬的血中尿酸降低作用。对比格犬(北山labes)强制口服给予了悬浮于0.5%甲基纤维素溶液中的试验化合物。在化合物给予前和给予4小时后,皮下给予了氧嗪酸钾(50mg/kg)。化合物给予8小时后,从桡侧皮静脉采血,之后分离了血浆。血中尿酸值使用LC-MS/MS法进行测定,通过下式求出了尿酸降低率。尿酸降低率(%)=(对照动物的尿酸值-给予试验化合物的动物的尿酸值)×100/对照动物的尿酸值给予8小时后,化合物(I)在10mg/kg的用量下显示出尿酸降低作用。由该结果显示出:化合物(I)对狗具有强力的血中尿酸降低效果。[参考例7]组织和血浆中的黄嘌呤氧化酶抑制活性的持续性对于本发明的“黄嘌呤氧化酶”,仅就本例而言,为了将氧化酶型的黄嘌呤氧化还原酶所承担的氧化反应催化活性、与氧化酶型和脱氢酶型两方的黄嘌呤氧化还原酶所承担的氧化反应催化活性进行区分,将前者称为“XO活性”,将后者称为“XOR活性”。对于“组织XO活性”、“血浆XO活性”、“组织XO抑制活性”、“组织XOR抑制活性”等,“XO活性”和“XOR活性”也具有同样的含义。“组织”包含肝脏、肾脏、脂肪组织、肠道、血管。需要说明的是,根据下述的结果,可以理解:对于本发明的化合物,在相同的样品中,XOR活性抑制率和XO活性抑制率为相同程度的数值。对于化合物(I),确认了组织XO活性、组织XOR活性和血浆XO活性。使用进食针(feedingneedle)对7~9周龄的Sprague-Dawley系雄性大鼠(日本CharlesRiver株式会社)强制给予了悬浮于0.5%甲基纤维素溶液中的试验化合物。给予后24小时或27小时后,进行了从腹部大动脉的采血和组织的取材。对所得的血液进行离心分离,采取了血浆。利用蝶呤由各型的黄嘌呤氧化还原酶氧化,生成作为荧光物质的异黄蝶呤的反应,测定了组织XO活性、组织XOR活性和血浆XO活性。将组织用包含1mMEDTA和蛋白酶抑制剂的pH7.4的磷酸钾溶液进行均质化,使各组织浓度达到肝脏:25mg/mL、肾脏:25mg/mL、脂肪:5mg/mL、肠道:5mg/mL、血管:30mg/mL,在4℃、12000rpm下离心了15分钟。在XO活性测定时,将组织均浆的上清液或血浆与包含50μM蝶呤的溶液混合,在37℃下进行了反应。在XOR活性测定时,将组织均浆的上清液与包含50μM蝶呤和50μM亚甲蓝的溶液混合,在37℃下进行了反应。作为对照,使包含氧化酶型黄嘌呤氧化还原酶(来自酪乳、Calbiochem公司制造)和50μM蝶呤的溶液利用同样的方法进行了反应。测定所生成的异黄蝶呤的荧光强度,利用对照的酶活性和蛋白浓度进行校正,以XO或XOR活性的形式进行了计算。XO抑制活性和XOR抑制活性利用下式求出。XO抑制活性(%)=(对照动物的XO活性或XOR活性-给予试验化合物的动物的XO活性或XOR活性)×100/对照动物的XO活性或XOR活性将给予约27小时后的组织和血浆XO抑制活性示于下表。[表1]组织和血浆中XO抑制活性(给予后约27小时解剖时)抑制的%(相对于媒介物)用量(mg/kg)110肝脏80%以上80%以上肾脏60%以上70%以上血浆25%以上40%以上关于化合物(I),在10mg/kg的用量下,将给予后27小时的肝脏的XO活性与对照动物进行比较,抑制了80%以上;将给予后27小时的肾脏的XO活性与对照动物进行比较,抑制了70%以上;将给予后27小时的血浆的XO活性与对照动物进行比较,抑制了40%以上。关于化合物(I),在1mg/kg的用量下,将给予后27小时的肝脏的XO活性与对照动物进行比较,抑制了80%以上;将给予后27小时的肾脏的XO活性与对照动物进行比较,抑制了60%以上;将给予后27小时的血浆的XO活性与对照动物进行比较,抑制了25%以上。另外,将给予24小时后的组织XO和XOR抑制活性示于下表。[表2]组织中XO和XOR抑制活性(给予后24小时解剖时)抑制的%(相对于媒介物)用量(mg/kg)110肝脏XOR80%以上80%以上肝脏XO80%以上80%以上肾脏XOR60%以上70%以上肾脏XO60%以上70%以上肠道XOR60%以上80%以上脂肪XOR30%以上60%以上血管XOR25%以上40%以上关于化合物(I),在10mg/kg的用量下,将给予后24小时的肝脏的XOR活性和XO活性分别与对照动物进行比较,抑制了80%以上;将给予后24小时的肾脏的XOR活性和XO活性分别与对照动物进行比较,抑制了70%以上;将给予后24小时的肠道的XOR活性与对照动物进行比较,抑制了80%以上;将给予后24小时的脂肪组织的XOR活性与对照动物进行比较,抑制了60%以上;将给予后24小时的血管的XOR活性与对照动物进行比较,抑制了40%以上。关于化合物(I),在1mg/kg的用量下,将给予后24小时的肝脏的XOR活性和XO活性分别与对照动物进行比较,抑制了80%以上;将给予后24小时的肾脏的XOR活性和XO活性分别与对照动物进行比较,抑制了60%以上;将给予后24小时的肠道XOR活性与对照动物进行比较,抑制了60%以上;将给予后24小时的脂肪组织XOR活性与对照动物进行比较,抑制了30%以上;将给予后24小时的血管XOR活性与对照动物进行比较,抑制了25%以上。由以上的结果显示出:本发明的化合物在各组织中具有长期、持续的XO活性、XOR活性抑制作用。产业上的可利用性本发明的化合物(I)的晶体和化合物(II)的盐及其晶体可用作药品。并且,这些化合物可用作药品制备用原料药。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
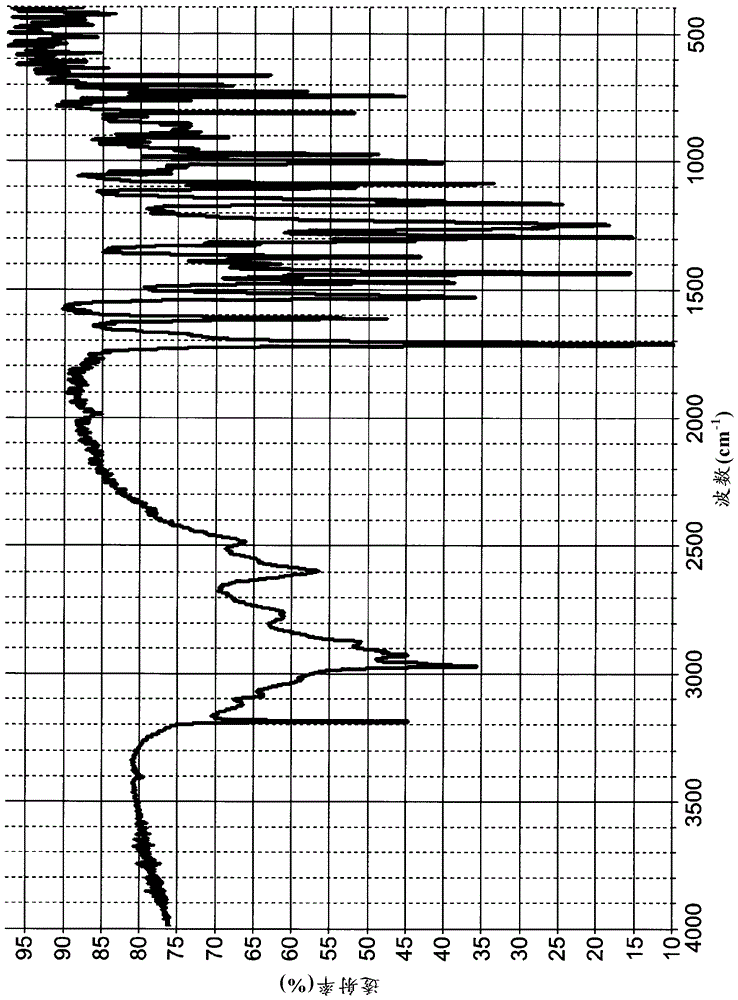
- 还没有人留言评论。精彩留言会获得点赞!
1
- 苯并二氮杂*衍生物的2-萘磺酸盐及晶型和它们的制备方法与制造工艺
- 一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法与制造工艺
- 苯并吲唑类衍生物及其制备方法、应用与制造工艺
- 一种二苯并环庚烷衍生物的制备方法与制造工艺
- Atropurpuran的苯并咪唑基和吗啉基衍生物组合物用于防治胰腺纤维化的制造方法与工艺
- 一种苯并咪唑衍生物镉配合物及其制备方法和应用
- 一种咪唑并吡啶巯乙酸类衍生物及其制备方法与应用
- Artalbic acid的O?(苯并咪唑基)乙基与O?(二羟乙胺基)乙基衍生物的组合物在抗病毒 ...的利记博彩app
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备抗骨质疏松药物的利记博彩app
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备防治胰腺纤维化药物的利记博彩app
- N-烯基苯并三唑类氮氧衍生物及其制备方法和应用
- 含有噻二唑衍生物、纳米铜和石墨烯的添加剂,添加剂制备方法,含该有该添加剂的润滑油的利记博彩app
- 一种具有抗菌活性的手性螺1,2,3-噻二唑衍生物及其合成方法和应用
- 含有噻二唑衍生物和纳米铜的润滑油添加剂,添加剂的制备方法,及含有该添加剂的润滑油的利记博彩app
- 作为免疫调节剂的1,3,4-*二唑和1,3,4-噻二唑衍生物的利记博彩app
- 含有噻二唑衍生物和石墨烯的润滑油添加剂,添加剂的制备方法,及含有该添加剂的润滑油的利记博彩app
- 一类1,3,4-噻二唑衍生物、制备方法及应用
- 一类1,3,4-噻二唑衍生物、合成方法及其应用
- 一种双磷酸酯唑类衍生物的制备方法
- 一种用于rv减速器的锂基润滑脂及其制备方法