制备作为可再充电电池阳极材料的MSnx纳米颗粒的方法与流程
本发明总体上涉及作为可再充电电池,尤其是钠离子或锂离子电池的阳极材料的MSnx纳米颗粒的制备方法,和包含此种材料的阳极的制备方法。
发明背景
尽管在最近几十年中对可再充电锂离子电池的材料已有广泛研究,但是石墨仍然是商业电池的最广泛使用的阳极材料。然而,石墨与许多合金型(例如Si、Ge、Sn)和转化型材料(例如Fe3O4、MoS2、SnSb)相比具有较低比容量和体积容量(372mAhg-1;820mAhcm-3)。虽然这些材料通常经历在锂化/脱锂化期间发生的大的体积变化,但是对于许多体系已经证实这种问题可以通过使用纳米结构化材料缓解。[1]虽然如此,此种大容量合金型或转化型阳极的工业化由于若干原因被阻碍。特别是对于转化型阳极,在超过1.0V vs.Li+/Li的电势下获得主要份额的容量,这导致相应全电池(full-cells)的能量密度低。其次,通常电池材料的合成太成本密集或太复杂而不能以工业规模执行。在作为能够替代商业电池中的石墨的现实备选的几种材料中包括Sn,原因在于它兼具大多数关键性能:高的体积容量和比容量(~7300mAhcm-3,992mAhg-1)、低的脱锂电势、高的导电性和合理的价格。事实上,当前在Sony的NexelionTM电池中使用基于无定形Sn-Co-C纳米复合材料的阳极,该电池已经引发对用于锂离子电池的Co-Sn基阳极的密集研究。[2]
因此,对于日益重要的应用例如便携式电子仪器或电动车,急迫需要替代石墨作为阳极的合适材料以改进可再充电电池尤其是锂离子电池的能量密度。
因此需要开发允许制备MSnx纳米颗粒的便宜且简单的程序,所述MSnx纳米颗粒作为可再充电电池,尤其是锂离子电池的阳极材料显示高的电化学性能。
技术实现要素:
因此,本发明的总体目的是提供制备MSnx纳米颗粒的方法,其中
M是选自Co、Mn、Fe、Ni、Cu、In、Al、Ge、Pb、Bi、Ga的元素,和
0<x≤10,优选0<x≤3
根据本发明,制备MSnx纳米颗粒的方法包括以下步骤:
-合成Sn纳米颗粒:用氢化物在无水极性溶剂中的溶液还原锡盐,将由所述溶液形成的固体Sn纳米颗粒分离和洗涤所述Sn纳米颗粒,
-合成M纳米颗粒:用氢化物在无水极性溶剂中的溶液还原金属盐,将由所述溶液形成的固体M纳米颗粒分离和洗涤所述M纳米颗粒,
-将所述Sn纳米颗粒和所述M纳米颗粒机械混合以将它们转化成MSnx纳米颗粒的纳米合金。
本发明的另一个目的是提供可再充电电池,尤其是钠离子或锂离子电池的阳极的制备方法,所述阳极包含通过本发明的方法获得的锡基材料。
优选地,所述机械混合步骤的M纳米颗粒和Sn纳米颗粒的摩尔比(M/Sn)可以在1:1-1:3范围内。
有利地,机械混合可以通过球磨达到,该球磨可以在空气或惰性气体中,例如在氮气下进行。优选地,球磨在空气中进行。
在一些优选的实施方案中,M是Co,x优选可以是大约2。
锡盐的还原反应优选在提高的反应温度下例如在50℃-70℃范围内的温度下进行。
金属盐的还原反应优选在更加提高的反应温度下例如在60℃-180℃范围内的温度下进行,这取决于金属盐的反应性。
适合的氢化物的实例是NaBH4、氢化锂、氢化钠、氢化钾、氢化镁、氢化钙、氢化三丁基锡、氢化二异丁基铝、氢化锂铝、三乙基硼氢化锂和它们的混合物。优选的氢化物是NaBH4。
无水极性溶剂的实例是1-甲基-2-吡咯烷酮(NMP),六甲基磷酰胺,1,3-二甲基-2-咪唑啉酮,1,3-二甲基-3,4,5,6-四氢-2(1H)-嘧啶酮,直链醚例如甘醇二甲醚、二甘醇二甲醚、三乙二醇二甲基醚,但不限于这些,亚砜例如二甲亚砜或环丁砜,但不限于这些,和它们的混合物。优选的无水极性溶剂是NMP。
适合的锡盐的实例是氯化锡、氟化锡、溴化锡、碘化锡、锡氧化物、锡硫化物、锡酸钠三水合物、四丁基锡和它们的混合物,优选氯化锡。
适合的合金化金属盐的实例是M氯化物(MCl2),M氟化物,M溴化物,M碘化物,M氧化物,M硫化物,M硫酸盐和它们的混合物,优选Co盐的混合物,最优选氯化Co(CoCl2)。
锡盐或金属盐的还原反应各自可以在惰性气体,优选在氮气下进行或也可以在空气中进行。
在优选的方法中,合成Sn纳米颗粒的步骤可以包括以下步骤:
-制备氢化物在无水溶剂中的溶液和锡盐在无水溶剂中的至少一种溶液,
-加热所述氢化物的溶液至所述锡盐的还原反应的反应温度,和
-当通过将所述一种或多种锡盐溶液快速注射到氢化物溶液中产生反应混合物而达到所述锡盐的还原反应的反应温度时,开始还原反应。快速意指添加以最高可能的速度并且无中断地进行。在反应混合物中,Sn纳米颗粒通过将一种或多种锡盐添加至氢化物中而生成,并且瞬时形成。
在另一个实施方案中,合成Sn纳米颗粒的步骤可以包括以下步骤:
-制备一种或多种锡盐在无水溶剂中的溶液和氢化物在无水溶剂中的至少一种溶液,
-加热所述锡盐的溶液至所述锡盐的还原反应的反应温度,和
-当通过将氢化物溶液快速注射到所述一种或多种锡盐的溶液中产生反应混合物而达到所述锡盐的还原反应的反应温度时,开始还原反应,其中Sn纳米颗粒瞬时形成。
在优选的方法中,合成M纳米颗粒的步骤可以包括以下步骤:
-制备氢化物在无水溶剂中的溶液和金属盐在无水溶剂中的至少一种溶液,
-加热所述氢化物的溶液至所述金属盐的还原反应的反应温度,和
-当通过将所述一种或多种金属盐的溶液快速注射到氢化物溶液中产生反应混合物而达到所述金属盐的还原反应的反应温度时,开始还原反应。快速意指添加以最高可能的速度并且无中断地进行。在反应混合物中,M纳米颗粒通过将一种或多种金属盐添加至氢化物中生成,并且瞬时形成。
在另一个实施方案中,合成M纳米颗粒的步骤可以包括以下步骤:
-制备一种或多种金属盐在无水溶剂中的溶液和氢化物在无水溶剂中的至少一种溶液,
-加热所述金属盐的溶液至所述金属盐的还原反应的反应温度,和
-当通过将氢化物溶液快速注射到所述一种或多种金属盐的溶液中产生反应混合物而达到所述金属盐的还原反应的反应温度时,开始还原反应,其中M纳米颗粒瞬时形成。
有利地,在注射后立即将已经在上述合成之一期间产生的反应混合物冷却至室温。更特别地,在一个优选的实施方案中,在相应地注射一种或多种锡盐溶液或一种或多种金属盐的溶液后立即将通过使相应的锡盐溶液或金属盐溶液与氢化物溶液合并形成的反应混合物冷却至室温,例如通过使用水冰浴进行冷却。
优选地,通过离心分离将形成的固体Sn纳米颗粒或M纳米颗粒从它们相应的反应混合物中分离。
然后相应地洗涤所获得的固体Sn纳米颗粒或M纳米颗粒,优选首先用溶剂如二甲亚砜(DMSO)然后用水洗涤。
在机械混合之前,可以在真空烘箱中在室温干燥Sn或M纳米颗粒。
本发明方法使用简单的制备程序,基于用于合成M-Sn基纳米颗粒的便宜前体,组合了湿化学合成和机械球磨。
可以通过将上述获得的MSnx纳米颗粒、炭黑、羧甲基纤维素(CMC)和水混合来制备阳极。然后将所获得的水性浆料涂覆在集电体上,随后在电池组装之前干燥。
使用此种阳极,可以根据本领域中熟知的程序制备锂离子电池或钠离子电池。
附图说明
当考虑本发明以下详细描述时,本发明将能更好地理解并且除上面提出的那些目的以外的目的也将变得清晰。该说明将参考附图,它们显示:
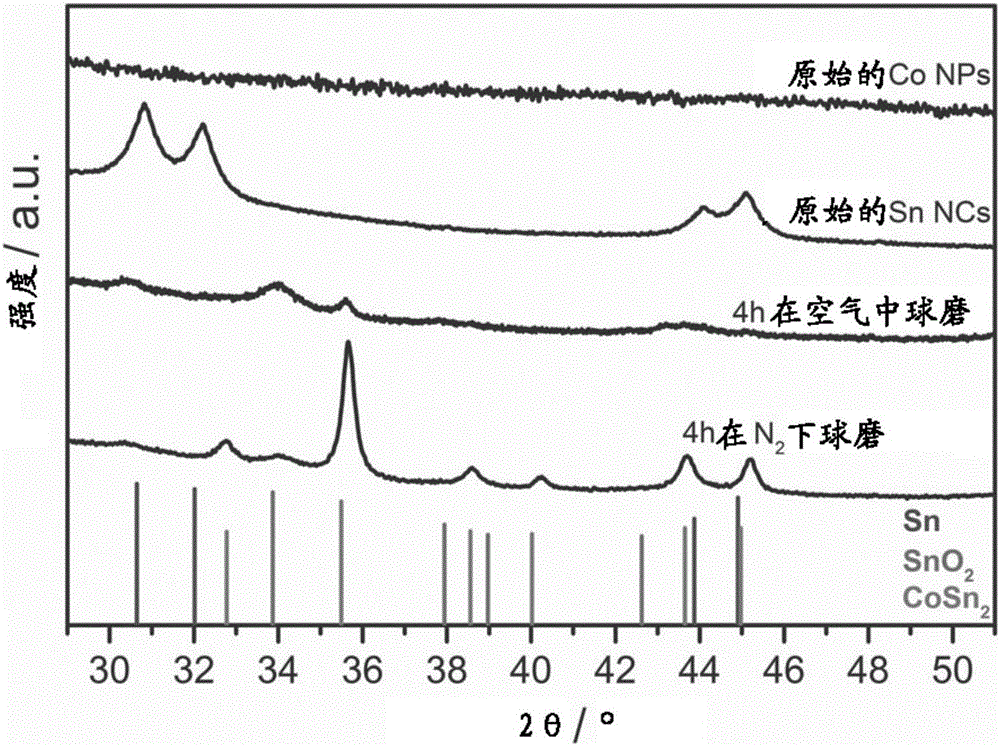
图1.通过在空气中球磨制备的Co NPs、Sn NCs、CoSnx NPs和通过本发明方法通过在氮气气氛下球磨制备的CoSn2 NCs的X射线衍射(XRD)图案。
图2.无定形Co NPs的EDX-谱。
图3.Co NPs(A)、Sn NCs(B)、CoSnx NPs(C)和CoSn2 NCs(D)的透射电子显微(TEM)图像。
图4.在锂离子半电池中在1984mAg-1的电流下在0.005-1.0V的电势范围内Sn NCs、CoSn2 NCs和CoSnx NPs的容量保持。
图5.对应于图4的Sn NCs(A)、CoSn2 NCs(B)和CoSnx NPs(C)的电流充电/放电曲线。
图6.在锂离子半电池中使用0.1Vs-1的扫描速率在0.005-1.0V电势范围中试验的Sn NCs(A)、CoSn2 NCs(B)和CoSnx NPs(C)的循环伏安图。
图7.在锂离子半电池中在0.005-1.0V电势范围内Sn NCs、CoSn2 NCs和CoSnx NPs的倍率性能(rate capability)测量。
图8.(A)用LiCoO2作为阴极材料的CoSnx NPs锂离子全电池在500mAg-1电流下的容量保持,和(B)与(A)对应的CoSnx/LiCoO2全电池的电流充电/放电曲线,以平均放电电压作为插入图(inset)。
具体实施方式
根据本发明方法,首先通过用氢化物在无水极性溶剂中的溶液还原相应的金属氯化物分开合成Sn纳米颗粒(NPs)和M NPs。
在Sn或M NPs,尤其是Co NPs的典型的合成中,将适合量的氢化物例如NaBH4溶解在合适量的无水极性溶剂例如1-甲基-2-吡咯烷酮(NMP)中,在搅拌的同时加热。为了合成Sn NPs,在达到所需的温度,例如60℃后,迅速地注射锡盐,例如SnCl2在无水溶剂例如NMP中的溶液。对于合成M NPs,在达到所需的温度,例如对于M=Co而言150℃或对于M=Mn、Fe、Ni而言120℃后,迅速地注射金属盐,例如CoCl2、MnCl2、FeCl2、NiCl2在无水溶剂例如NMP中的溶液。
立即形成固体Sn NPs或M NPs。在注射后,将各自的悬浮液冷却至室温,例如用水冰浴冷却。通过离心分离使相应获得的材料从它们的溶液中分离并用二甲亚砜(DMSO)洗涤一次和用水洗涤两次以除去未反应的NaBH4和水溶性副产物例如NaCl。最后可以在真空烘箱中在室温下干燥相应的反应产物。通常,反应按如下量得到所需产物:对于Sn NPs 67%,对于Co NPs 98%,对于Fe NPs 36%,对于Mn NPs 16%和对于Ni NPs 34%。
在上述方法中,以下化学物质适当地通用于获得Sn NPs或M NPs。然而,为了考虑MCl2和SnCl2的不同反应性,使用不同的反应温度和前体浓度:
I.作为除NMP以外的无水溶剂:
任何酰胺例如六甲基磷酰胺、1,3-二甲基-2-咪唑啉酮或1,3-二甲基-3,4,5,6-四氢-2(1H)-嘧啶酮。
II.作为NaBH4的替代:
任何碱金属或碱土金属氢化物例如氢化锂、氢化钠、氢化钾、氢化镁、氢化钙或其它金属氢化物例如氢化三丁基锡、氢化二异丁基铝、氢化锂铝或三乙基硼氢化锂。
III.作为SnCl2的替代:
任何锡卤化物例如氟化锡、溴化锡、碘化锡;任何锡氧化物、锡硫化物、锡酸钠三水合物或四丁基锡。
IV.作为MCl2的替代:
任何金属卤化物例如金属氟化物、金属溴化物、金属碘化物;任何金属氧化物、金属硫化物或金属硫酸盐。
上述化学物质可以单独使用或与它们相应I-IV组中的一种或多种其它成员组合使用。
颗粒尺寸可能受数种特性例如氢化物量、反应温度和冷却速度的影响。
取决于采用的NaBH4的量,NPs的尺寸可以改变,即NaBH4的量越高,颗粒越小。另外,为了以高产率制备小尺寸的NPs,需要过量的NaBH4。
Sn NPs或M NPs也可以在室温下制备,然而,较小NPs的形成很可能在高温下是有利的。
可以如下获得具有大致5-10nm的直径的Sn NPs:采用大约96倍过量的NaBH4作为还原剂并在SnCl2的注射后直接地急速冷却。
可以如下获得具有大致4-10nm的直径的M NPs:采用大约4-8倍过量的NaBH4作为还原剂并在MCl2的注射后直接地急速冷却。
可以通过本领域普通技术人员已知的任何适当的冷却技术实现急速冷却。
另外,已经发现,Sn或M NPs的此种合成可以在空气中进行,这显著地降低成本(材料以及工作时间)。
除容易进行和相当便宜之外,制备本发明中使用的Sn NPs和M NPs的方法与文献中描述的方法相比具有若干优点。这种合成程序的优点以下:
I.不需要使用表面活性剂。
II.反应也可以在空气中进行。
III.不贵且安全的化学物质:这里优选使用的NaBH4是最便宜的金属氢化物,
IV.洗涤程序:所获得的Sn或M NPs简单地用水在空气中洗涤。
V.取决于所使用的NaBH4的过量和反应温度,可以调整颗粒的尺寸。
在合成Sn NPs和M NPs后,制备Sn NPs和M NPs的混合物。这些混合物中的M纳米颗粒和Sn纳米颗粒的摩尔比在1:1至1:3范围内。
在空气中或在氮气下球磨Sn NPs和M NPs的混合物,目标是将材料合金化,获得MSnx纳米颗粒。有利地,以1800-2400rpm的频率将M纳米颗粒和Sn纳米颗粒球磨2-4小时。
可以通过将MSnx NPs、炭黑、CMC和水混合,优选通过使用球磨机混合例如1h制备阳极。然后将所获得的水性浆料涂覆在集电体如Cu集电体上,随后在电池组装之前干燥,例如在80℃下在真空下干燥过夜。
实验部分
I.所使用的材料
化学物质和溶剂:氯化锡SnCl2(99.9%,Alfa Aesar)、CoCl2(98%,Sigma-Aldrich)、1-甲基-2-吡咯烷酮(NMP,无水,99.5%,Fisher BioReagents)。
电池组分:炭黑(CB,Super C65,由TIMCAL提供),羧甲基纤维素(CMC,品级:2200,Daicel Fine Chem Ltd.);氟代碳酸亚乙酯(FEC,Solvay,电池级),LiPF6在碳酸亚乙酯/碳酸二甲基酯中的1M溶液(EC:DMC:1:1,Merck,电池级),玻璃微纤维隔片(GF/D,Whatman,Cu箔(9μm,MTI Corporation)
II.方法
CoSnx NPs的合成
实施例1:CoSn2纳米晶体(NCs)的合成
根据本发明,CoSn2 NCs的合成包括合成Sn NCs,合成Co NPs,和通过球磨Sn NCs和Co NPs合成CoSn2 NCs:
合成Sn NCs:
在Sn NCs的典型合成中,在氮气下将96mmol NaBH4溶解在85mL无水NMP中并加热到60℃,同时机械搅拌。在达到60℃后,迅速地注射1mmol SnCl2预先溶解在无水NMP中的溶液并使用水冰浴立即将该反应混合物冷却至室温。通过离心分离从溶液分离反应产物,用二甲亚砜洗涤一次,然后用水洗涤两次。获得Sn NCs。
反应产量是80mg(67%)。所获得的产物的XRD图案仅显示对应于结晶β-Sn的峰(图1,索引到四方晶系Sn,空间群I41/amd(141),ICDD PDF条目号:00-004-0673)。反应产物的TEM分析显示颗粒是多分散的,具有5-10nm的尺寸(图3B)。
合成Co NPs:
在Co NPs的典型合成中,使用与Sn NCs相似的程序,但有改变,尤其是反应温度和前体浓度):在氮气下将32mmol NaBH4溶解在15mL无水NMP中并加热到150℃,同时机械搅拌。在达到150℃后,迅速地注射8mmol CoCl2预先溶解在无水NMP中的溶液并使用水冰浴立即将该反应混合物冷却至室温。通过离心分离从溶液分离反应产物,用二甲亚砜洗涤一次,然后用水洗涤两次。获得Co NPs。
反应产量是460mg(98%)。所获得的产物的XRD图案显示获得无定形的Co NPs。图2示出了无定形Co NPs的EDX-谱。对应于S(样品的大约1wt%)的峰可能归因于残留DMSO。反应产物的TEM分析显示颗粒是多分散的,具有4-7nm范围的尺寸(图3A)。
合成CoSn2纳米晶体(NCs):
为了制备Co-Sn基NCs,以30s-1的频率将如上制备的1.4mmol Sn NCs与如上制备的0.7mmol Co NPs球磨4小时。在氮气气氛下加载用于球磨的烧杯并密封。
反应产量是200mg(96%)。所获得的产物的XRD图案显示在惰性条件下球磨导致形成结晶的CoSn2纳米合金(参考图案:四方晶系SnO2,空间群P42/mnm(136),ICDD PDF条目00-077-0448;四方晶系CoSn2,空间群I4/mcm(140),ICDD PDF条目00-025-0256)。反应产物的TEM分析显示颗粒是多分散的,具有6-20nm的尺寸(图3D)。
实施例2:合成CoSnx纳米颗粒(NPs)
为了制备Co-Sn基NCs,以30s-1的频率将如上制备的1.4mmol Sn NCs与如上制备的0.7mmol Co NPs球磨4小时。在空气中加载用于球磨的烧杯。
反应产量是200mg(96%)。所获得的产物的XRD图案显示在空气中球磨导致形成无定形的CoSnx NPs。样品的主要级分无定形化,仅具有在34°对应于SnO2和在35.5°对应于CoSn2的小特征。反应产物的TEM分析显示颗粒是多分散的,具有6-20nm的尺寸(图3C)。
III.制备Co-Sn基电极、电池组装和电化学测量
为了电极制备,通过以500rpm球磨1小时将相应的NPs(64wt.%)与CB(21wt%)、CMC(15wt%)和作为溶剂的水混合来制备水性浆料。将所得的浆料涂覆到铜集电体上,在电池组装之前在真空下在80℃干燥12小时。在于Ar填充的手套箱(O2<0.1ppm,H2O<0.1ppm)中组装的气密性硬币型电池中进行电化学测量,对于锂离子半电池试验使用元素锂或对于锂离子全电池试验使用在铝箔(MTI)上的LiCoO2。使用一片玻璃微纤维作为隔片。作为电解质,使用在具有3%FEC的EC:DMC中的1M LiPF6。将FEC添加到电解质中以改进循环稳定性。电流循环试验在室温下在MPG2多路工作站(BioLogic)上进行。通过用于半电池和全电池试验的Co-Sn纳米颗粒的质量(不包括CB和粘结剂)将容量标准化。
IV.表征
使用碳涂覆的Cu栅格作为基底(Ted-Pella)用Philips CM30 TEM显微镜在300kV下获得透射电子显微(TEM)图像。使用NanoSEM 230进行能量分散X射线光谱(EDX)测量。在STOE STADI P粉末X射线衍射仪上测量粉末X射线衍射(XRD)。
V.电化学结果
图4示出了Co-Sn基NPs在0.005–1.0V电势范围中在1984mAg-1的高电流下经1500个循环的容量保持。1984mAg-1的电流对于Sn而言相当于2C倍率,基于形成Li4.4Sn的992mAhg-1的理论容量。假定Co不贡献于容量,则CoSnx NPs和CoSn2 NCs的理论容量为795mAhg-1,因此与纯Sn相比更低。对于恒电流循环试验,上限截止(cut-off)电势限于1.0V,从而仅包括与全电池中的高能量密度对应的过程。如可从图4中在1984mAg-1下的恒电流循环看出的那样,在首次100个循环期间在容量快速增加后,Sn NCs以及CoSnx NPs和CoSn2 NCs显示~570mAhg-1的容量。然而Sn NCs在400个循环后显示明显的容量衰减,Co-Sn基NPs显示好得多的容量保持。特别地,对于CoSn2 NCs,在1500个循环后,仍然保持462mAhg-1。对于CoSnxNPs,容量保持甚至更好,为525mAhg-1,这相当于在1984mAg-1的电流下经1500个循环仅8%的衰减。因此,通过本发明方法获得的CoSnx NPs具有超高的循环稳定性。包括Co的CoSnxNPs与纯Sn NCs相比的优异循环稳定性可能归因于两个作用。由于Co不形成锂合金这一事实,它可以在循环期间充当非活性基体并因此缓冲由Sn的锂化/脱锂化引起的体积变化。此外,Co的存在可以防止Sn NCs聚集并因此进一步改进循环稳定性。CoSn2 NCs和CoSnx NPs之间在循环稳定性方面的差异可能归因于这样的事实:CoSnx NPs由于它们通过球磨在空气中制备而更加氧化。更高含量的氧化物可能导致形成更加有效的Li2O基体以缓冲体积变化和抑制Sn域在循环期间的烧结。应该指出的是,对于所有三个体系,在第一放电循环由于固体电解质形成而获得~30%的值之后,平均库仑效率在循环期间是99.6%。Sn NCs、CoSn2NCs和CoSnx NPs的平均脱锂电势同样低,在循环期间具有~0.5V vs.Li+/Li的稳定值(图5A至5C和图6A至6C)。
为了评价Co-Sn基NPs的倍率性能,进行在0.2C-10C之间的电流倍率下的恒电流循环试验(图7,1C=992mAg-1)。由于在所有情况下NPs的小尺寸和因此增强的反应动力学,观察到类似的好的倍率性能,不过对于0.5C–2C的电流,Sn NCs显示与CoSn2 NCs相比容量低~50mAhg-1。对于高达10C的倍率,所有三种材料仍然保持~350mAhg-1的容量。引起兴趣地,可以观察到,在如此高电流下,容量在循环期间增加,导致在所述倍率逐步降回到0.2C期间获得相同或甚至更高的容量。特别是对于CoSnx NPs,初始在0.5C–2C的倍率下观察到的在容量方面与CoSn2 NCs的微小差异在循环期间完全减小。
为了试验Co-Sn基NPs在更加实际的条件下的应用性,进行使用LiCoO2作为阴极的阳极限制的全电池试验。选择CoSnx NPs作为阳极材料,原因在于它们与Sn和CoSn2 NCs相比优异的容量保持。这里,所有容量和电流与CoSnx NPs的质量有关。最初将CoSnx NPs/LiCoO2的全电池充电到2000mAhg-1以考虑在第一循环中的不可逆充电损失。对于随后的循环,充电和放电限于500mAhg-1。在这些条件下以500mAg-1的电流循环,CoSnx NPs显示稳定的容量,对于50个循环具有3.2V的平均放电电压。基于阳极容量和放电电压,大致估计CoSnx NPs的比能量密度与石墨相当(372mAhg-1,3.6V vs.LiCoO2)。然而,考虑到本体β-Sn(~7.3gcm-3)和Co(~8.9gcm-3)与石墨(~2.2gcm-3)相比高得多的密度,使用CoSnx NPs可潜在地提高体积能量密度高达4倍。
图8示出了用LiCoO2作为阴极材料的CoSnx NPs锂离子全电池的电化学性能:图8A:在500mAg-1电流下的容量保持。图8B:与(A)对应的SnSb/LiCoO2全电池的恒电流充电/放电曲线,以平均放电电压作为插入图(inset)。限制充电和放电容量到500mAhg-1的情况下循环电池。显示的容量和电流与CoSnx NPs的质量有关。
总之,本发明的方法允许通过用在NMP中的NaBH4简单还原相应的金属氯化物合成具有直径≤10nm的Co NPs和Sn NCs,随后通过球磨将它们转化成金属间结晶和无定形的Co-Sn纳米合金。尽管CoSn2 NCs显示对于数百循环的好的循环稳定性,但是无定形的CoSnxNPs显示优异的容量保持,在1984mAg-1下经1500个循环仅有8%衰减。另外,用LiCoO2作为阴极材料在锂离子全电池中试验,CoSnx NPs提供500mAhg-1的稳定容量,平均放电电压为3.2V。考虑到便宜和容易大规模化的制备方法和它们的由高可循环性以及高体积和比能量密度表征的优异电化学性能,这里提供的CoSnx NPs作为Li离子和Na离子电池的高性能阳极材料具有巨大潜能。
虽然已显示和描述了本发明目前的优选实施方案,但是应清楚地理解,本发明不限于它们,而是可以在权利要求书范围内以其它方式多样地实施和实践。
参考文献
[1]a)P.G.Bruce,B.Scrosati,J.-M.Tarascon,Angew.Chem.Int.Ed.2008,47,2930-2946;b)M.F.Oszajca,M.I.Bodnarchuk,M.V.Kovalenko,Chem.Mater.2014,26,5422-5432。
[2]a)A.D.W.Todd,R.E.Mar,J.R.Dahn,J.Electrochem.Soc.2007,154,A597-A604;b)X.-L.Wang,W.-Q.Han,J.Chen,J.Graetz,ACS Appl.Mater.Interfaces 2010,2,1548-1551。
- 还没有人留言评论。精彩留言会获得点赞!